|
Authors: Lidón Carretero Vilarroig & Belén Gómez Giménez Epigenetics in ART“Epigenetics explains why different cells perform different tasks despite their same DNA content” 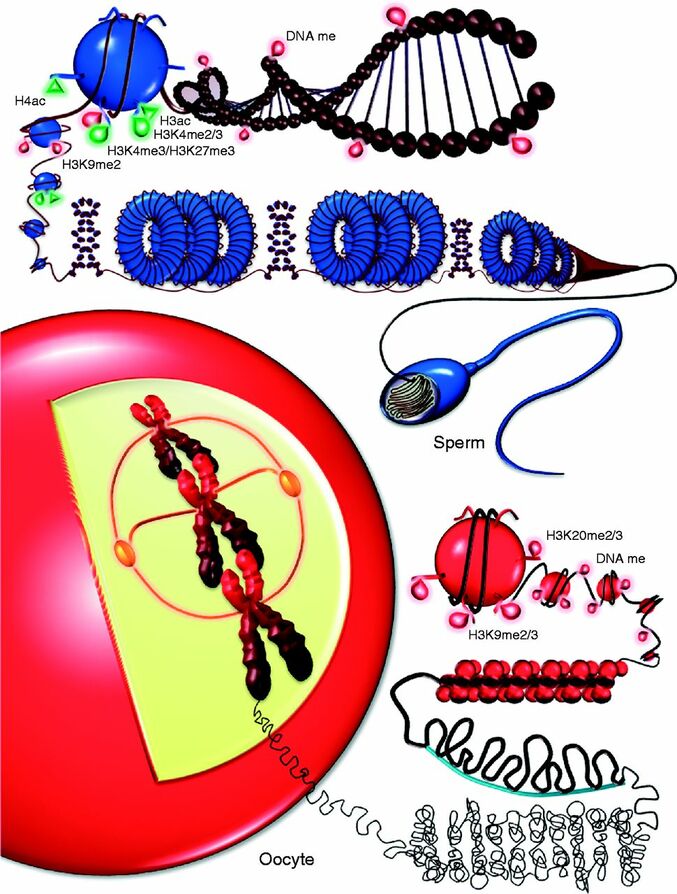 Figure 1. Epigenetic landscape in gametes (1). INTRODUCTION In vitro fertilisation (IVF) techniques have gained popularity over the years. Since the implantation of IVF techniques in 1978 more than 8 million babies have been born thanks to artificial reproductive technology (ART) worldwide (2). Nonetheless, there exists certain evidence that these techniques may induce long-term consequences on the health of the offspring (3). Consequently, exogenous hormonal stimulation, embryo culture media or embryo manipulation are currently being studied as factors affecting IVF babies. One of the mechanisms by which IVF can affect long-term health outcomes is epigenetics. This refers to modifications that affect genetic expression without altering the actual DNA sequence. The epigenome is responsible for expressing or repressing genes needed for differentiation between cellular types. Some of these epigenetic mechanisms include:
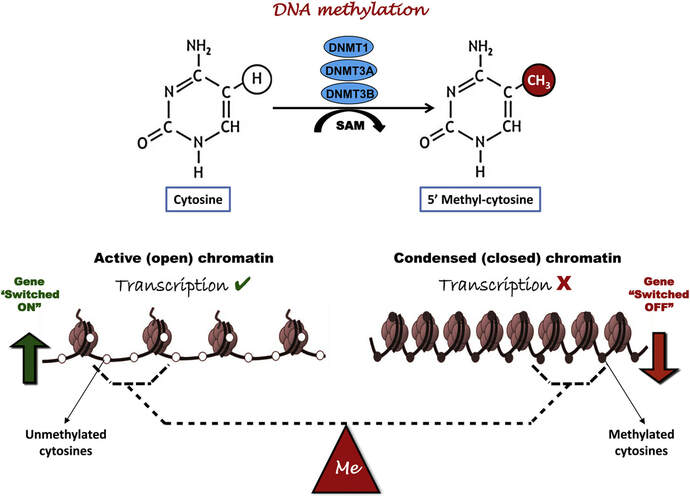 Figure 2. Methylation means the addition of a methyl group (S-adenosylmethionine (SAM)) to the C-5 position of a cytosine molecule. This transference is catalyzed by methyltransferases (DNMTs). Methylation results in chromatin condensation, thus switching off gene transcription (4). Epigenetic changes begin during gametogenesis (F1), when methylation patterns that differ between male and female gametes are established (imprinting) (Fig. 3). After fertilisation, when both gametes come together to form a zygote (F2), the embryo genome is reprogrammed, meaning every gene is de-methylated and given a new methylation pattern, with the exception of those imprinted. This is important as both gametes, oocyte and spermatozoon, are highly specialised cells with a gene expression profile suitable to their very own function. Thus, they must be reprogrammed in order to acquire pluripotency for subsequent cell division and differentiation. These changes are completed by implantation (5). Here we aim to summarize different epigenetic mechanisms in the sperm and the oocyte, as well as to review the possible effects of ART on the embryo epigenome. 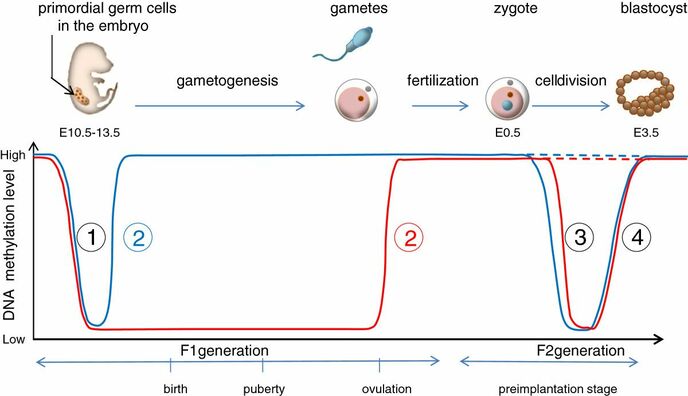 Figure 3. Reprogramming entails removal or resetting of the most previous epigenetic marks in order to allow cells to specialise and differentiate. (1) First DNA demethylation occurs in the male (blue curve) or female (red curve) primordial germ cells of F1. (2) Then, until puberty, de novo methylation takes place in the genome of the gametes. Maternal methylation marks are established at a later stage (ovulation) than paternal marks. (3) Demethylation occurs again after fertilisation in the F2 zygote. Nonetheless, paternal and maternal imprinted genes maintain their methylation pattern (dotted curves). This allows for the inheritance of parent-specific monoallelic expression in F2 somatic cells. (4) Methylation changes are completed by implantation. Modified from (5). EPIGENETICS IN THE SPERM CELL During spermiogenesis, the majority of sperm histones are substituted by protamines. Protamines are small arginine-rich proteins typical of mammals, which compact the sperm genome at a higher level than histones, thus forming a toroidal structure (Fig. 4). This structure allows for better motility of the sperm, as well as protection against exogenous DNA damage within the female tract. There are two types of protamines in humans: P1 and P2. Both work together in a 1:1 ratio in order to form the toroidal structure. Alterations of this ratio make DNA more accessible, thus increasing exposure to exogenous and endogenous sources of damage, too (6). Moreover, this imbalance is related to higher implantation and fertilisation failure rates (7). Although protamine substitution is of extreme importance for proper sperm morphology and function, about 5% of histones remain exposed to modifications. Histone-linked DNA sequences exhibit a specific pattern of modifications aimed to activate or repress marks in the promoters of genes related to imprinting and embryogenesis (8, 9). For instance, trimethylated lysine 4 on histone H3 (H3K4me3) is characteristic of developmental promoters, regions containing homeotic genes of the HOX family, some ncRNAs and paternally expressed imprinted loci (10). Alterations in some epigenetic marks such as PTMs have also been associated with fertility problems. Schon et al have recently published a study seeking alterations in histones PTMs in sperm samples (11). This study included samples from 31 men with normal or abnormal semen parameters. Their findings suggested that those with altered semen parameters displayed a decrease in histone 4 acetylation, as well as alterations in the methylation profile of H3K9 and H4K20. 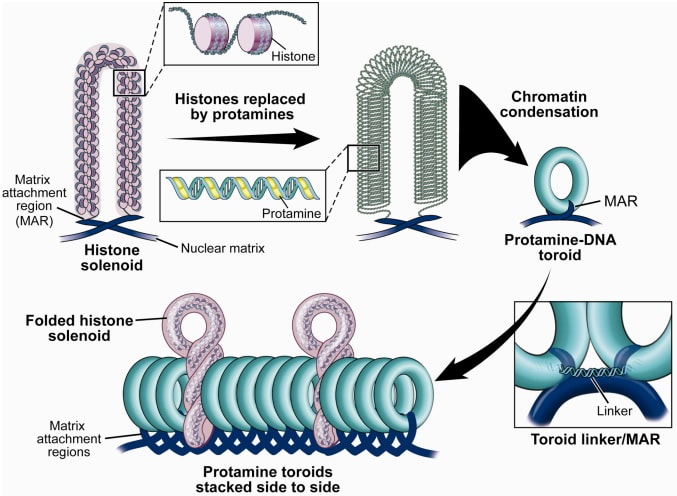 Figure 4. Sperm DNA organization. Histones are substituted by protamines P1 and P2, forming a toroidal structure. Such toroids maximize DNA compaction protecting DNA from endogenous damage. A smaller amount of DNA is kept associated with histones present in the sperm nucleus, with the remaining DNA attached to the nuclear matrix at the so-called Matrix Attachment Regions (MARs). Modified from (12). Regarding imprinting, one of the most extensively studied paternally-imprinted genes is H19. This gene is located at the long arm of chromosome 11 (11p15.5) and encodes a ncRNA involved in body weight regulation and cell multiplication (13). H19 expression is associated with another gene named Insulin Like Growth Factor 2 (IGF2). Whereas H19 acts as a tumour suppressor, IGF2 is a very important growth factor related to embryonic development, and both share common enhancers downstream of H19 (14) (Fig. 5). Demethylation of H19 activates the maternal allele, which represses IGF2; simultaneously the paternal allele is repressed because of the methylation state, thus promoting expression of IGF2 (15). Defects in H19 methylation have been detected in different cohorts of infertile men (16). Epigenetics plays an important role in fertility, but also in other phenotypic characteristics through parents to offspring. Nevertheless, the mechanisms involved in those phenotypic traits are still not well understood. PTMs are hard to study due to protamine substitution and tight packaging. For example, some authors have associated nutritional status and physical activity levels with epigenetic changes in somatic cells (17, 18), although the sperm epigenome can also become modified due to environmental factors. Recently, a link between sperm epigenome and obesity and bariatric surgery was published (19). This study showed that sperm cells from men with higher body mass index (BMI) (BMI = 31.8) have a different epigenetic profile than those with normal BMI (BMI=22.9) (19). Differences were detected in the methylation profile (methylome), as well as in the small ncRNA expression profile, whereas histone position was not altered. These results could offer an explanation as to why children from obese fathers are more likely to suffer from metabolic diseases, regardless of the mother’s weight (20, 21). 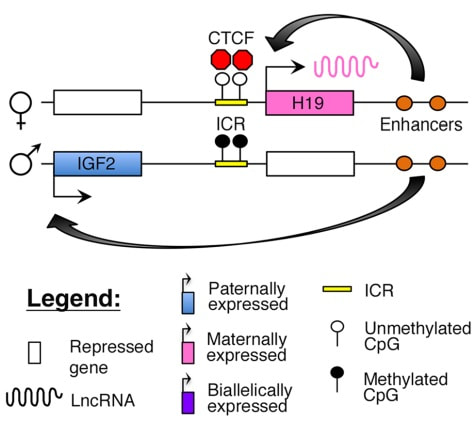 Figure 5. Proposed model of imprinting at the H19–IGF2 locus. Imprinting control region (ICR) in the H19-IGF2 locus is unmethylated in the maternal, preventing access of the IGF2 promoter, thus allowing H19 expression. On the other hand, ICR in the H19-IGF2 paternal allele is methylated, causing the repression of H19 and activation the expression of IGF2 (14). EPIGENETICS IN OOCYTES Several families of proteins contain enzymes that are responsible for maintaining the correct methylation pattern required in a particular developmental environment, such as the oocyte (22). Oocyte methylation is laid down during follicle development (1). These enzymes include, for instance, DNA methyltransferases (DNMT), demethylases (TET), histone acetyltransferases (HAT) or histone deacetylases (HDAC). As outlined above, several genes are subjected to imprinting, and their differential expression patterns depend on the epigenetic mark of each allele, which is specific for each of the parentals (23). Oocyte DNA, as opposed to sperm DNA, is compacted with histones. Research on animal models has shown that imprinted genes become methylated at different stages of folliculogenesis (24). When primordial follicles are activated, acquisition of maternal DNA methylation begins and it is completed in metaphase II (MII) oocytes (Fig. 6) (25). In mice, it is well known that maternal imprinted gene acquisition is gradual during folliculogenesis. It starts at primordial and primary follicle stages, and complete methylation occurs in the antral-ovulated follicles (24, 25). As an example, Snrpn gene is a maternally imprinted gene whose methylation begins at first stages of the follicle development. It is involved in the Prader-Willi syndrome in humans, caused if maternal methylation is not correctly established. During later stages of follicle development, PEG3 and IGFR2 genes acquire their methylation mark. Their tasks involve proliferation and intracellular trafficking. Up to early antral follicle stages, PEG1/Mest genes get their methylation pattern, playing an important role in foetal development. Finally, when follicles become antral-ovulatory follicles Impact gene becomes methylated (24, 25). This is a translational regulator that ensures constant high levels of translation upon a variety of stress conditions. Thus, similar results were observed during human oocyte growth: methylation of the SNRPN human imprinted allele starts during the germinal vesicle and metaphase I stages, and it is completed at MII stage (25). As aforementioned, the H19 gene is paternally imprinted. Its methylation is established during spermatogenesis, so the H19 allele is methylated before the maternal allele in embryos. (24). In contrast, SNRPN is methylated on both the paternal and the maternal allele but, as Lucifero et al (2004) observed, maternally imprinted alleles become methylated before the paternal ones (Fig. 6) (25). 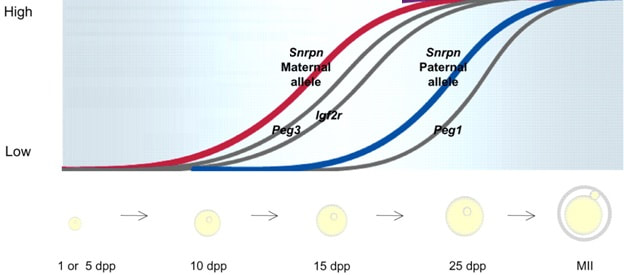 Figure 6. Schematic representation of the acquisition of the methylation profile of both paternal and maternal alleles of Peg3, Igf2r, Snrpn and Peg1 genes during oogenesis (25). EPIGENETICS IN EMBRYOS To guarantee the correct embryo genetic methylation following fertilisation, a new epigenetic reprogramming occurs again during early embryo development. Both pronuclear genomes are demethylated; by the four-cell stage the embryo genome is activated and a new increase in methylation is observed until blastocyst stage. During embryo development paternal and maternal genomes possess asymmetric epigenetic modifications (1). Uysal et al (2017) correlated demethylation and methylation patterns during oocyte maturation and early embryo development with DNMT expression (Fig. 7) (26). 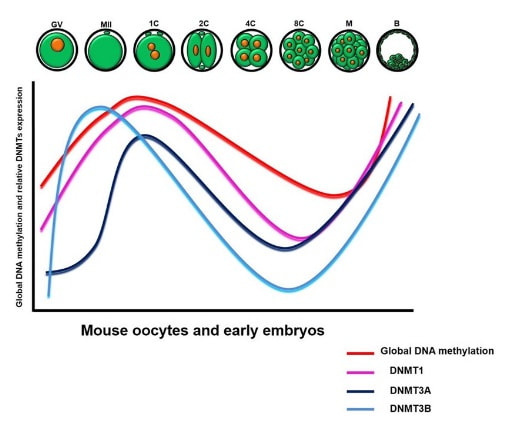 Figure 7. Global DNA methylation profile and relative expression levels of DNMT1, DNMT3A and DNMT3B proteins in the mouse oocytes and early embryos. GV: germinal vesicle oocyte, MII: metaphase II oocyte, 1C: 1-cell embryo, 2C: 2-cell embryo, 4C: 4-cell embryo, 8C: 8-cell embryo, M: morula, B: blastocyst (26). ABNORMAL EPIGENETIC CHANGES IN ART ART involves hormone stimulation, IVF or intracytoplasmic sperm injection (ICSI), embryo culture and cryopreservation among others. Even though ART is globally applied and well-established, several studies have shown association of ART with an increased incidence rate of certain imprinting disorders such as Beckwith–Wiedemann, Angelman, Prader-Willi and Russell-Silver syndromes (27, 28). ART procedures are applied during a window of important epigenetic reprogramming, gamete maturation and preimplantation embryo development (27). Furthermore, gametes and embryos are exposed to in vitro conditions, and although these aim to mimic the actual physiological conditions, it is still not clear how closely they match the in vivo environment. So, it has been suggested that these non-physiological conditions can increase the risk of developing imprinting disorders (27, 28). Several studies on animal models have suggested epigenetic changes in both gametes and embryos originated following ART. For instance, superovulation protocols have been found to result in altered levels of enzymes from the DNMT family during early embryo development in mice (26, 29). Market-Velker et al (2010) further observed demethylation of maternally imprinted genes (Snrpn, Peg3, Kcnq1ot1) and methylation of paternally imprinted genes (H19) in mouse embryos after superovulation (30). Similarly, Velker et al (2017) detected demethylation of Peg1/Mest gene during in vitro embryo culture in mice (31). Research lines focused on sheep and cattle have also observed that in vitro culture of oocytes and embryos could alter methylation and expression of imprinted genes. These observations led to the conclusion that gamete manipulation and/or ART may actually be responsible for what had been described as the large offspring syndrome (LOS), ruminant version of the Beckwith–Wiedemann syndrome (BWS) in humans, with similar phenotypical manifestations (32). As for vitrification, Ghazifard et al (2019) have recently observed increased levels of HAT and acetylation of histone 4 (acH4K12) in mouse oocytes, in comparison to fresh ones (33). These data add up to those reported by previous studies in which in vitro maturation was shown to disturb the expression of histone acetyltransferases or histone deacetylases in oocytes or embryo development (34, 35). Human studies on micromanipulation and epigenetics are hard to perform given how valuable and scarce samples are and because clinical cohort studies are extremely difficult and randomized prospective trials are impossible. Despite such obstacles, some findings have revealed an association between several imprinting disorders and ART (36). And even though studies have reported a three-fold to six-fold increase in the occurrence of BWS in association with ART when compared to the general population, abnormal DNA methylation has not been able to be consistently identified in IVF children (22). On the contrary, a recent study showed that ART manipulation after controlled ovarian stimulation does not increase the risk of abnormal expression and DNA methylation of imprinted genes (H19, SNRPN and IGF2) (37). It must be taken into consideration that some infertility problems of couples resorting to ART are related to advanced maternal age; so, it would be feasible to argue that epigenetic abnormalities may actually arise from the underlying cause of infertility rather that the treatment itself (38). Furthermore, these studies have been performed using poor-quality embryos that were unsuitable for transfer. This is why some authors have suggested that the poor quality of the samples could be the reason behind the increase of imprinting disorders and not ART itself exclusively (38). However, no clear affirmation can be stated yet regarding clear and evident causes for any of the above-summarised consequences. CONCLUSIONS The majority of the epigenetic reprogramming occurs during gametogenesis and embryo development, coinciding in time with most ART procedures. In spite of the millions of babies born through ART around the world and even though these approaches are currently considered a secure technology, several studies on both animals and humans suggest that non-physiological conditions may in fact induce aberrant epigenetic reprogramming, abnormal development and imprinting disorders. However, due to limitations in the availability of human samples, difficulty to create proper control gametes and the fact that most of these studies analyzed poor-quality embryos, a consistent relationship between an increased risk of imprinting disorders and ART exposure has not yet been properly demonstrated. Moreover, some authors suggest that a potential increase could be attributed to infertility rather than ART procedures (32, 39). Therefore, it seems reasonable to think that additional studies with non-infertile couples and good-quality embryos would be necessary to examine whether there exists an actual link between imprinting disorders and potential long-term effects of ART procedures. REFERENCES
1 Comment
Non-invasive PGTAuthors: Lidón Carretero Vilarroig & Sara Gómez García "These are exciting and challenging times, and non-invasive PGT may be one step closer to becoming a reality" 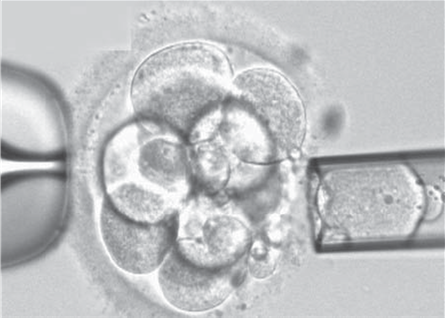 Figure 1. Biopsy of a human embryo. The holding pipette on the left abuts the zona pellucida of the embryo. The biopsy pipette on the right is inside the opening in the zona pellucida. One cell is inside the pipette (1). INTRODUCTION The importance of aneuploidy screening in assisted reproduction has gained popularity since the introduction of preimplantation genetic testing (PGT) at the beginning of the 1990s (2). PGT for aneuploidies screening (PGT-A) is recommended for couples experiencing recurrent pregnancy loss and implantation failure, since aneuploidies could be the reason behind these issues. Aneuploidy rate increases with maternal age (3), which is why PGT-A is also offered to women above 37 years old. A few years ago, infertility was only attributed to women, but now it is well documented that male factor plays also an important role (4), and so, aneuploidies in sperm or chromosome alterations as a result of defective meiosis in sperm are too indications to perform PGT. Different techniques have been developed in order to select euploid embryos for transfer. Initially, fluorescent in situ hybridization (FISH) was the only technique to test for aneuploidy, but it only allowed for the assessment of some chromosomes. Subsequently, Comparative Genomic Hybridization array (CGHa) and most recently Next-Generation Sequencing (NGS) enable the detection of cytogenetic changes, including every chromosome in the complement. In order to perform PGT, embryo biopsying is needed. Different techniques are available to collect the required sample: biopsy of polar bodies or embryo at either cleavage or blastocyst stage are the most extended, yet invasive methodologies. A new approach for non-invasive PGT (NIPGT) is reviewed below: the analysis of cell-free DNA from embryo culture. CURRENT PGT TECHNIQUES 1. Polar Body (PB) Biopsy First polar body and second polar body are indicative of oocyte maturation (oogenesis). Once the primary oocyte completes the first meiotic division, first polar body is produced, whereas the second one is only formed if fertilization is achieved (as a result of the completion of the second meiotic division). During oocyte and embryo development polar bodies degenerate after being extruded. However, they can be collected during an in vitro fertilization cycle, either sequentially or simultaneously (Figure 2). 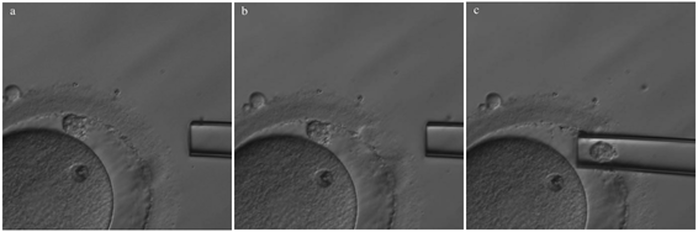 Figure 2. Laser-assisted polar body (PB) biopsy. Prior to biopsy the oocyte is carefully positioned with the first PB in focus. (a) An opening of approximately 20 μm is formed in the zona pellucida (ZP) by two laser shots (14 ms pulse duration). (b) The biopsy pipette can be easily introduced through the opening for aspiration of the PB. (c) Finally, the PB is aspirated through the pipette. A skilled operator can carry out the whole procedure in less than 1 min. Modified from (5). It is important to point out that both PBs contain only maternal genetic material. Thus, paternal contribution cannot be analyzed, which is the main limitation of this technique. Nevertheless, PB biopsy is less invasive than other methodologies, and as such it avoids ethical, religious and legal problems related with embryo manipulation. 2. Embryo biopsy at cleavage stage Three days after fertilization, the embryo reaches cleavage stage. At this point, a normal embryo should contain around 16 individually distinguishable cells known as blastomeres. Biopsy at cleavage stage implies the removal of one or two blastomeres. Similarly to PB biopsy, a hole in the zona pellucida (ZP) is created using either a laser pulse or acid Tyrode's solution (Figure 3). The blastomeres are pulled away using a biopsy pipette (6) and collected into a microtube. The genetic material of the blastomere(s) is then amplified, and the result is considered representative of the embryo genome. In 1990 Hardy et al concluded that this practice does not compromise in vitro development (7). Biopsy of cleavage stage embryos was performed in approximately 90% of all reported PGT cases in 2012 (8), but it does not allow to distinguish mosaic embryos (9). These are embryos with two or more cell lines containing both euploid and aneuploid cells. Mosaic embryos can be considered for transfer following specific recommendations (Preimplantation Genetic Diagnosis International Society (PGDIS)). 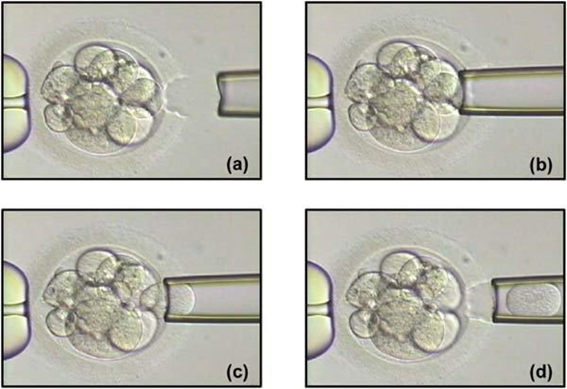 Figure 3. Cleavage-stage embryo biopsy. (a) Cleavage-stage embryo ready to be biopsied. (b) The embryo is immobilized with a holding pipette and one blastomere is selected. (c-d) The nucleus-containing targeted blastomere is gently aspirated into the pipette (6). 3. Embryo biopsy at blastocyst stage The embryo reaches blastocyst stage on day 5 of embryonic development or day 6 after fertilization. Membrane ion transporters and channels, such as Na+/K+ pump, are activated and so fluid is accumulated in the blastocoel (Blastocoel Fluid, BF). This process is termed cavitation, and two structures are at this point differentiated: the inner cell mass (ICM), which will eventually form the embryo, and the trophectoderm (TE), which will give rise to the placenta and associated tissues. Since the first blastocyst biopsies until now, PGT cases have increased on Day 5 or Day 6 (D5/D6) embryos. This consists in making a small hole, between 30 to 35 µm, in the ZP with the ICM positioned either at 8 o’clock or 11 o’clock, this is, away from the laser. This provides direct access to TE cells, so they can be biopsied with no damage to the ICM (10). Similarly to PB and cleavage stage embryo biopsy, the ZP can be breached by mechanical or physical methods, laser being the most common one (although more cells are removed by this procedure ~5-10 cells). Furthermore, it can be performed at two different times (10, 11):
Not only D5 biopsy does not compromise embryo viability, but it also enables a high pregnancy rate (60-69.2%, approximately). This methodology allows to reduce both technical errors and the risk of mistakenly detecting mosaicism, because it implies the removal of 5-10 cells (12, 13). 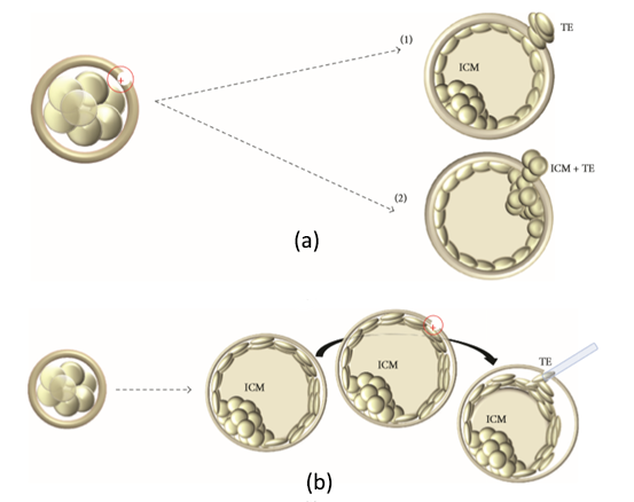 Figure 4. Two different stages to make a hole in the zona pellucida (ZP). (a) The hole is made on D3 and by D5, as the blastocyst expands, and either trophectoderm (TE) cells (a1) or inner cell mass (ICM) and TE cells (a2) will protrude from the ZP. (B) Both hole and biopsy are carried out on day 5 or 6. Modified from (11). NON-INVASIVE PGT 1. Evolution of PGT towards non-invasive PGT (NIPGT) Although embryo biopsy is considered to be a safe procedure, other less invasive methods are being investigated as an alternative to provide lesser manipulation of the embryo. Recent studies have shown that BF and culture medium could contain small amounts of cell-free embryonic DNA, which may be used for PGT (14, 15, 16). Currently, there are three ongoing research lines on the topic:
2. What is the origin of BF-DNA? The origin of BF-DNA is not entirely clear, but certain studies indicate it may arise from cells undergoing apoptosis during blastulation as part of normal development, lysed cells or even discarded abnormal cells (17). As discussed later, several studies have investigated whether such DNA may correspond to DNA from TE (see the following sections “Blastocoel fluid”, “Cell free DNA on the spent culture media” and “Blastocoel fluid and culture”). 2.3. Blastocoel fluid (BF) In 2012, a study published by Alessandro et al (18) detected a series of metabolites in the BF. The methodology employed, referred to as blastocentesis, consisted in immobilizing expanded blastocysts using a holding pipette in a plate without culture medium in order to avoid contamination, and then using an intracytoplasmic sperm injection (ICSI) pipette to aspirate the BF for analysis (Figure 5). Blastocentesis is nowadays a routine methodology employed by professionals in some laboratories to collapse blastocysts before vitrification. 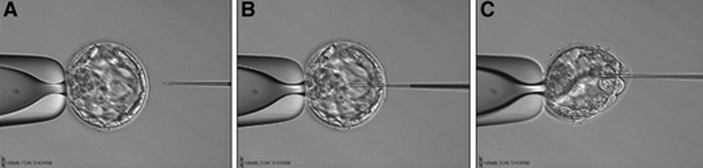 Figure 5. Biopsy of blastocoel fluid. (A) The intracytoplasmic sperm injection (ICSI) pipette is inserted inside the embryo blastocoel. (B) The blastocoel fluid (BF) is aspirated through the ICSI pipette. (C) BF-aspirated embryo collapses (19). From these findings, Pallini and collaborators performed the blastocentesis technique in order to find DNA in BF (14). This was the first study exploring the presence of genetic material in BF, and DNA was detected in 90% of the samples. Furthermore, the authors used the DNA found to confirm the sex of embryos as well as to detect aneuploidies. Several investigations have been conducted with the aim of detecting DNA in BF samples. First of all, DNA from BF or TE is amplified and then analysed. In these studies, rate of amplification failure in TE samples was found to be lower than 2%; however, amplification of DNA from BF was harder, with higher failure rate than DNA from TE (Table 1). 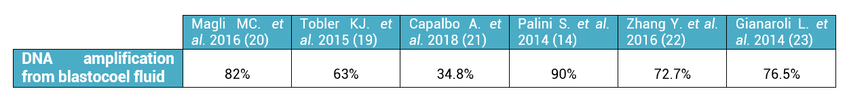 Table 1. DNA amplification from blastocoel fluid (BF). Different studies detected DNA from blastocoel fluid and obtained amplification rate lower than that from trophectoderm (TE) samples. Even so, the quantity of DNA obtained from BF was similar to that obtained from a single blastomere (14, 22). Conversely, Li and co-authors have recently reported that DNA extracted from BF is insufficient for amplification and sequencing (25). Table 2 exhibits a relation of studies with concordant and discordant results between BF and TE samples, respectively (Table 2). 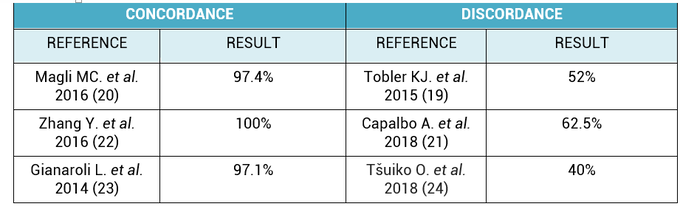 Table 2. Different studies indicated concordance between blastocoel fluid (BF) and trophectoderm (TE) cells, which are in contrast with the second column, showing studies that reported discrepancies in the results. Consequently, further investigations will be in order. 4. Cell-free DNA on spent culture media The presence of embryonic free cells in the spent embryo culture have been shown by several studies. The origin of cell-free DNA is still under investigation, but the most likely source is remnants of apoptotic embryo cells (17). A study by Stigliani et al about mitochondrial DNA content in embryo culture medium showed that 99% of embryos at day 2 an day 3 were accompanied by free DNA in their spent culture media (15). The authors additionally reported two important things: (i) a larger amount of free DNA in the medium of bad-quality cleavage embryos, and (ii) higher mitochondrial/genomic DNA ratios in spent medium were associated with successful implantation outcomes. Since the discovery of the presence of free DNA in embryo culture media, some researchers have focused on evaluating the concordance between cytogenetic results of invasive techniques and the analysis of free DNA. In order to accomplish such goal, embryos are cultured individually and researchers collect a microdrop of media after 3, 4 or 5 days of culture. Simultaneously, embryo biopsy is performed and both samples are analyzed. Results reported suggested that cell-free DNA analysis is a promising option for NIPGT. Feichtinger et al (26) found a 72.2% concordance between results from PB biopsies and culture media using CGHa. Xu et al (27) in turn analysed the genetic complement of free DNA from culture media, as well as DNA from TE biopsies by NGS. Their results showed a specificity of 84% (false positives) and a sensitivity of 88.2% (false negatives). However, this technique would need further refinement, since more recent findings have suggested that spent culture media contains not only embryonic, but also maternal DNA, which is a confounding factor. In fact, Vera-Rodriguez and collaborators (17) reported a concordance of only 30.4% between culture media DNA and TE DNA when analysed by NGS. This could be attributed to maternal contamination, as well as to the inclusion of mosaic embryos in the study, which may actually hinder reliable diagnosis. Such big differences between findings may be attributed to the lack of a standardized protocol to develop NIPGT. Culture systems and conditions, culture volume, as well as DNA amplification methodology, differ between research groups, and so it becomes necessary to find reproducible conditions to verify the consistency of such results. 5. Embryo culture medium and blastocoel fluid (ECB) In 2018, preliminary studies were carried out to combine DNA from blastocoel fluid and culture medium (ECB) and, thereby increase embryonic DNA amount to improve accuracy and reliability of the non-invasive preimplantation genetic screening. Fully expanded blastocysts were collapsed with a laser (16) or a laser was used to breach the ZP (25), thus obtaining enough DNA from the mix-up of spent culture medium and BF. Data reported by Li et al (25) after amplification and sequencing of DNA from both ECB and TE have shown different aneuploidy chromosomal patterns detected in approximately 50% of the cases. Conversely, a 100% of embryonic DNA has been successfully amplified by the team of Kuznyetsov and collaborators (16). These authors have shown the concordance rate between TE biopsy and the combination of BF and culture medium to be 87.5% for whole chromosome copy number. Other sources of DNA must be carefully considered in order to analyse data, since foreign DNA may eventually reach the media. Contamination with non-embryonic DNA (either maternal or paternal DNA) (17, 21, 28) or degraded DNA fragments from culture medium needs to be avoided. One way to minimise such contamination is by transferring embryos to fresh medium on day 4, which poses no particular problem if the goal does not include to measure other molecules such as metabolites, for instance. COMPARISON BETWEEN INVASIVE PGT AND NON-INVASIVE PGT As we have already explained, there are different methodologies to get a small sample of the embryo in order to perfomr PGT. Table 3 sumarizes advantages and disadvantages of each one of them. Polar body biopsy is minimally invasive and avoids ethical and legal issues, but only allows to analyze maternal genetic complement. On the other hand, biopsy at day 3 and at day 5 are more invasive but enable the detection of the whole genetic complement. Non invasive techniques such as blastocoel fluid, spent media culture or ECB are still under development, but promises minimal manipulation. 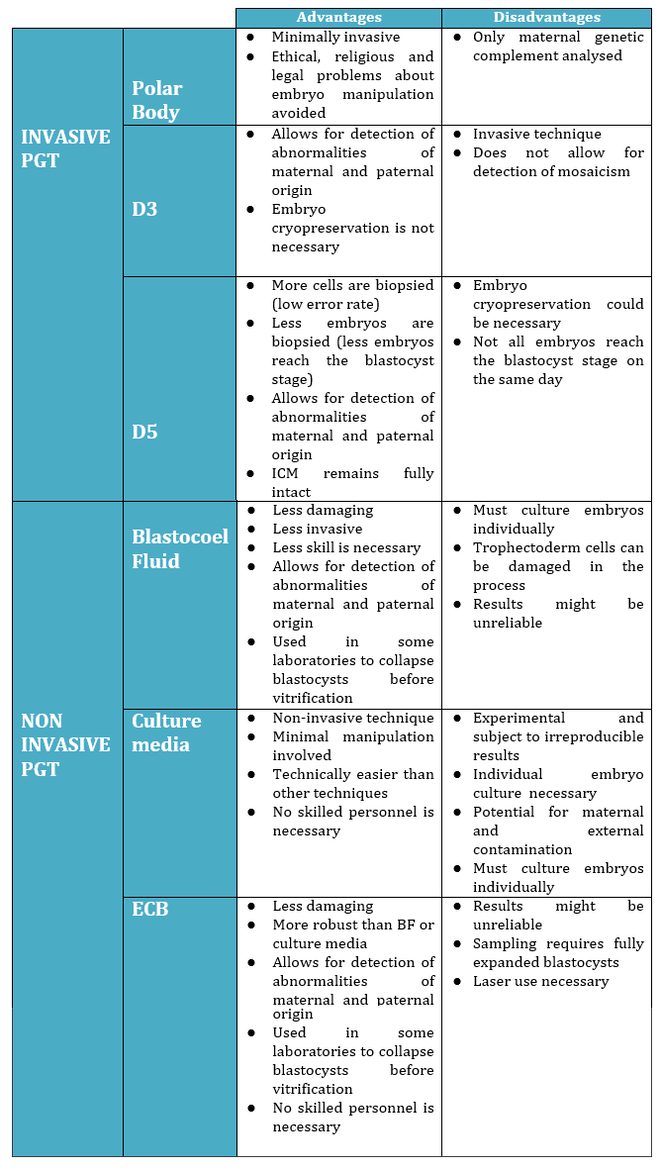 Table 3. Advantages and dissadvantages of each methodology. CONCLUSIONS Further research is still necessary to improve NIPGT until reliable consistent results are obtained, as well as efficiency concordance and similar amplification failure as that of current PGT. However, before implementation of NIPGT, several aspects must be kept in mind:
Given current available data, blastocoel fluid, culture spent media or both are potential candidates to become the next sources of embryonic DNA. This could eventually revolutionise the horizons of PGT and the achievable clinical outcomes. Technical hindrances are, as usual, in the way of improving and implementing actual routine applications of new methodologies. But, these are exciting and challenging times, and non-invasive PGT may be one step closer to becoming a reality. REFERENCES:
Authors: María Caballero Sastre & Raquel Pillado González “Currently, performing any cryopreservation technique results in some degree of damage to the sperm.“ 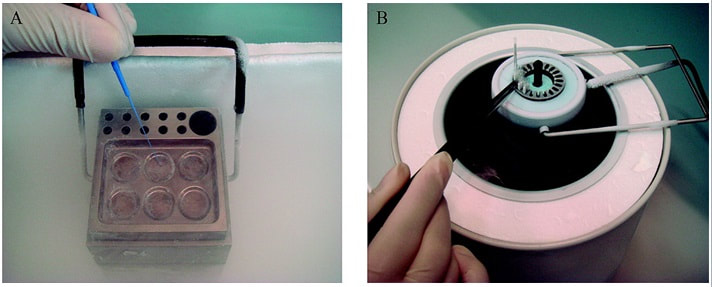 Figure 1. Solid Surface Vitrification (A) and slow cooling equipment (B) (1). THE HISTORY OF SPERM CRYOPRESERVATION Human semen cryopreservation has a long history that begins at the end of the nineteenth century. After prior observations on sperm surviving cooling at very low temperatures (-150º C) (2), Mantegazza (1866) first suggested the idea of human sperm banks (3). Years later, Mantegazza proved it was possible to extend human sperm lifespan up to four days by cooling at moderate sub-zero temperatures (-17º C) (4). Although a few extra days of storability does not make much difference in practical terms, this was the starting point of further research aimed to develop techniques that would allow for longer storage periods. The next leap forward in this field was the discovery of the cryoprotectant properties of glycerol in 1949. This molecule proved to be an effective cryoprotectant agent (CPA) when combined with bovine sperm, and allowed for the development of new cryopreservation methods through which sperm could preserve their motility and fertilization capabilities even after the freezing-thawing process (5). However, during the following years this new technique was primarily applied to cryopreservation of farm animal semen rather than human semen (6). In 1953, at the University of Iowa, the first case of live birth from cryopreserved sperm was reported (7). This successful birth, along with reports of high survival percentage (67%) of human sperm after cryopreservation, popularized the utilization of glycerol with human semen. The most common method to use glycerol was the protocol described by Bunge and colleagues, which stated the processing of sperm in a 10%-glycerol solution before freezing it with dry ice (7). A decade later, the use of liquid nitrogen was introduced for long-term sperm cryopreservation. This new method led to the progressive normalization of sperm freezing as a widespread practice in healthcare (8). With the availability of long-term storage and the extended use of sperm freezing, new methods and variants were developed over time, such as slow freezing, fast freezing, LN2 vapours or lyophilization, which will be later described. Nowadays, cryopreservation is routinely used in most assisted reproduction centres for numerous reasons:
It is important to possess a clear understanding of the semen cryopreservation process due to its current importance in clinical and research environments. Modern assisted reproduction practices are unthinkable without this tool. CRYOPRESERVATION TECHNIQUES Before being frozen, a sperm sample needs to be appropriately processed in order to separate sperm cells from the seminal plasma. This helps increase the concentration of high quality spermatozoa for a later use. Different techniques for sperm selection have been reviewed in our previous post. Cryoprotectants The process of cryopreservation may involve irreversible cellular damage due to a change in the osmotic balance (11). Upon freezing of the extracellular water, the solute concentration increases in this fraction. As a result, the intracellular water is transported out of the cell to restore the osmotic balance, which may lead to cell dehydration and irreversible membrane damage (12, 13). Consequently, CPAs need to be incorporated along the sperm sample following processing. These molecules will protect spermatozoa by reducing intracellular ice formation and by decreasing the freezing point of the medium and the solute concentration present in the extracellular environment (14). There are two kinds of cryoprotective agents: permeable and non-permeable. The permeable ones, such as glycerol, dimethyl sulfoxide or ethylene glycol, protect the intracellular structures and biomolecules. Non-permeable agents, like sucrose, polysaccharides and some proteins, contribute to keeping the osmotic equilibrium, thus preventing cellular dehydration (11). Procedures - Cryopreservation in liquid nitrogen (LN2) This is the most commonly used method to cryopreserve sperm. It can be subdivided into three methods:
- Cryopreservation in microdroplets Microdroplets are sometimes used in the clinic to cryopreserve sperm in small volumes. This is preferred in cases such as epididymal sperm aspiration. Around 50-100 μL of the sperm-CPA solution are placed on a dry ice plate up to freezing (Fig. 2); droplets formed in such a way are then kept in vials and plunged into LN2 (18).  Figure 2. Cryopreservation in droplets. Dry ice plate used to cool down samples that form sperm droplets (in yellow). - Vitrification This technique consists of cooling the sample at ultrarapid rates, so that the water solidifies (vitrifies) as a glass-like structure rather than forming ice crystals (19). However, even though vitrification should cause minimal damage, this is not always the case in clinical practice. One of the most frequent problems is the requirement for very high CPA concentrations that sperm do not tolerate well (20). Despite this, some studies have demonstrated it is possible to perform vitrification without using CPAs (20-22). Also, the large volume of sperm typically used impedes the cooling of the sample at the appropriate speed, causing ice formation (4). Despite these limitations, a vast proportion of clinics use vitrification as a routine practice due to its practical advantages. In recent years, a new variant of this technique has been developed. This evolved version is called Solid Surface Vitrification (SSV) (23, 24). For this procedure, the sample or tissue is directly exposed to a metal surface previously precooled at -160º C before the use of LN2 (Fig. 1). This method prevents the apparition of nitrogen bubbles and evaporation that would slow the cooling rate (24). This technique has been previously applied to animal mature oocytes and human gametes and embryos, yielding successful results (23, 24). OUTCOME COMPARATIVE BETWEEN TECHNIQUES Because every cryopreservation technique shows different advantages and disadvantages, it is important to bear in mind how sperm will be affected during the thawing process. Parameters such as motility, viability, morphology and DNA integrity are evaluated accordingly. Regarding LN2 techniques, no studies so far have directly compared post-thaw sperm quality following slow and rapid freezing. The literature shows agreement between studies on the main problem in both processes, which is controlling cooling rates (reviewed in 16). If the cooling rate is too fast, ice crystals may be formed inside the cells. By contrast, if it is too slow, the result could be cell contraction due to osmotic stress (25). This issue can be faced by using an automated programmable freezer, but only when keeping a large number of samples (26). Even though several reports have compared fast and slow freezing in animal reproduction, conclusions are controversial. Some studies in semen from horse (27) and buffalo (28) have reported better results when using fast thawing, whereas other authors have found no difference between fast and slow thawing rates (29). Nevertheless, it seems clear that the critical point lays on thawing matching the freezing process. Considering rapid freezing, thawing is recommended to be also carried out at a fast rate to avoid formation of intracellular ice crystals. Likewise, for the slow-rate cooling procedure, the sample needs a slow thawing protocol, since cells need more time to rehydrate (30). On the other hand, all studies on the use of LN2 show similar or slightly better results regarding the aforementioned parameters when using nitrogen vapours (31-33). These results, however, are obtained after short-term storage of samples in nitrogen vapours of up to three months. When stored for longer, sperm quality decreases. Consequently, this method is only recommended for short-term storage (further research would be needed in order to support its application for long-term storage) (34). Upon comparison between vitrification and LN2 techniques, different results can be highlighted. Certain authors determined that results of sperm parameters such as motility, viability and normal morphology were similar between vitrification and rapid freezing techniques (21). On the contrary, different results were found for DNA fragmentation rates. Whilst some groups found that DNA fragmentation was significantly higher for the rapid freezing technique (12, 21) or for LN2 vapours (24), other groups obtained contradictory or uncertain results (35, 36). For instance, DNA fragmentation has been observed to increase over time when analyzing semen 6h after thawing, compared to recently-thawed samples (35). Despite the different results obtained, vitrification shows important advantages compared to other available techniques. Some of these advantages are: 1) unnecessary use of CPAs; 2) the technique is simpler and faster compared to conventional slow freezing, due to the fact that once the sample is kept in a proper container (such as cryoloop or straws), it is rapidly plunging into LN2 to be stored; 3) no requirement for programmable freezers; 4) the sample is free of seminal plasma and potential pathogens (vitrification is usually performed after swim-up); 5) no requirement for post-thaw processing (reviewed in 6). TRENDS IN IMPROVING SEMEN CRYOPRESERVATION Currently, performing any cryopreservation technique results in some degree of damage to the sperm (4). The severity may differ depending on the initial quality of the sample, being greater in poor quality semen. Luckily, the application of ICSI allows for the successful use of low quality sperm (if necessary) even after having been cryopreserved. Side effects of cryopreservation on sperm include reduced motility, vitality, viability and increased DNA damage. Although motility is the most affected parameter, DNA damage entails greater detrimental effects regarding embryo viability (6). The majority of the harm produced by cryopreservation occurs during the freezing and thawing phases, the crucial moments being between -15º C and -60º C. It is worth mentioning that a considerable damage is produced by CPAs themselves; these agents cause oxidative stress that derives in the formation of reactive oxygen species (ROS) (8, 10, 37). Furthermore, these components affect the polyunsaturated fatty acids in plasmatic membranes due to lipid peroxidation (10). Consequently, phospholipids reservoirs such as egg yolk are usually added to the freezing media. As a more direct countermeasure, several current research lines look into numerous antioxidant components in order to be added to freezing media, thus avoiding the damage caused by ROS. Examples of these antioxidants are TAT-peroxiredoxin-2 fusion protein, quercetin or melatonin. Preliminary research indicates that their presence results in higher motility and viability rates post-thawing, along with a reduction of intracellular ROS levels (8, 10). Other approaches consider the utilization of protocols that may directly disregard the use of CPAs, such as certain vitrification protocols previously mentioned. Sperm freezing entails other associated problems, too, such as the loss of chromatin and acrosome integrity that had been observed post-thaw. Recent data have reported differences in the levels of DNA and acrosome integrity after cryopreservation depending on the freezing technique used (24). The difference in DNA integrity levels is suspected to be due to the cold shock faced by the samples (24). This issue could be amended by the development of media able to preserve sperm without freezing, an avenue that is currently being pursued. For instance, Riel and colleagues have reported that the use of an electrolyte-free medium for short-time (1 week) storage of semen yields better levels of DNA integrity in comparison to traditional cryopreservation. If the storage period capacity could be further improved, this might become a rather attractive alternative (38). Lyophilization or freeze drying is an experimental technique that has been proven less harmful to the DNA (4, 10, 38). In order to perform this method, the sample must be cooled below the triple point of water (Fig. 3). At this temperature solid water (ice) sublimates when the pressure is decreased and exits the cell, leaving it fully dehydrated (4, 10). However, this process irreversibly damages the sperm membrane, thus resulting in non-motile or even non-viable (dead) sperm. Nevertheless, studies on mouse sperm have shown that lyophilized spermatozoa can be used for fertilization with the assistance of ICSI (39). Although the first attempts to use lyophilization on human sperm were in the 50s, today there is still a lacking protocol for this technique that is able to preserve both sperm motility and viability (4). 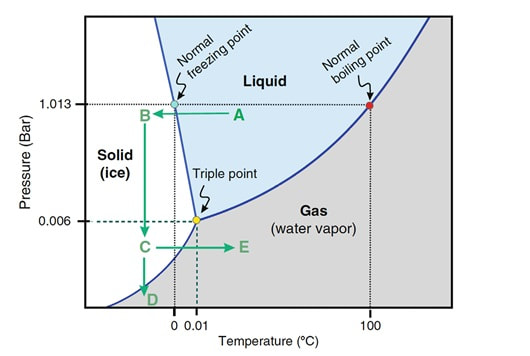 Figure 3. Water phase diagram showing the relation of the conditions of temperature and pressure for freeze-drying (not in scale). Samples are frozen by reducing the temperature (A to B) and then the pressure is also reduced by aspiration (creating vacuum) so the sample lies below the ‘triple’ point (C) for both temperature and pressure (this is the point where all states co-exist). From here on the sample is subjected to a controlled increase of temperature or to a further decrease of pressure to sublimate ice (for detailed current sperm freeze-drying protocols, see (40) and (41)). Modified from (41). The main advantages of freeze drying are: the possibility to preserve spermatozoa with high DNA integrity for at least a year and a half (39), the inactivation of viruses that may be present and the fact that liquid nitrogen is not required. Additionally, samples can be stored at 4º C and transported at room temperature (4). To date, this method still remains experimental regarding humans, due to the lack of actual data on the matter (4, 10). CONCLUSIONS Cryopreservation has gone a long way. Its use in reproductive medicine got to revolutionize the horizon for infertile couples. New doors opened decades ago, and it is fair to reason new ones will open in the near future. Egg donation, social freezing, embryo cryopreservation. Times and timing have changed for patients, and clinics and reproduction centres faced the need for evolution in order to cope with rising approaches. In spite of the variety of options for semen cryopreservation, all of them present their own limitations. Continuous research allows for the discovery of new ways to correct these flaws; however, there lies a long path ahead, and further studies will be required before any improvement can be incorporated to routine practice. REFERENCES
1. Wang X, Catt S, Pangestu M and Temple-Smith P. Live offspring from vitrified blastocysts derived from fresh and cryopreserved ovarian tissue grafts of adult mice. Soc Reprod Fert.2009;138(3): 527–535. 2. Varghese AC, Nandi P, Mahfouz R, Athayde KS, Agarwal A. Human Sperm Cryopreservation. In: Varghese, AC., Nandi, P., Mahfouz, R., Athayde, KS., Agarwal A, editor. ANDROLOGY LABORATORY MANUAL [Internet]. Cleveland Clinic. 2014; p.196–206. 3. Bunge, GR., & Sherman, KJ. Fertilizing Capacity of Frozen Human Spermatozoa. Nature.1953; 172(4382):767-8. 4. Mocé E, Fajardo AJ, Graham JK. Human sperm cryopreservation. EMJ. 2016;1:86–91. 5. Polge C, Smith AV, and Parkes AS. Revival of Spermatozoa after Vitrification and Dehydration at Low Temperatures Nature.1949; 164, 666. 6. Sharma R, Kattoor AJ, Ghulmiyyah J, Agarwal A, Sharma R, Kattoor AJ, et al. Effect of sperm storage and selection techniques on sperm parameters. Syst Biol Reprod Med. 2015;61(1):1–12. 7. Bunge RG, Sherman JK. Fertilizing capacity of frozen human spermatozoa. Nature. 1953;172:767–768. doi: 10.1038/172767b0. 8. Rozati H, Handley T, Jayasena C. Process and Pitfalls of Sperm Cryopreservation. J Clin Med. 2017;6(89):1–13. 9. Tiwari A, Tekcan M, Sati L, Murk W, Stronk J. A new media without animal component for sperm cryopreservation : motility and various attributes affecting paternal contribution of sperm. J Assist Reprod Genet. Journal of Assisted Reproduction and Genetics; 2017;34:647–57. 10. Karimfar MH, Niazvand F, Haghani K, Ghafourian S, Shirazi R, Bakhtiyari S. The protective effects of melatonin against cryopreservation-induced oxidative stress in human sperm. Int J Immunopathol Pharmacol. 2015;28(1):69–76. 11. Sieme H, Oldenhof H, Wolkers WF. Mode of action of cryoprotectants for sperm preservation. Anim Reprod Sci [Internet]. Elsevier B.V.; 2016;1–14. 12. Elliott GD, Wang S, Fuller BJ. Cryobiology Cryoprotectants : A review of the actions and applications of cryoprotective solutes that modulate cell recovery from ultra-low temperatures. Cryobiology [Internet]. Elsevier Ltd; 2017;76:74–91. 13. Royere D, Barthelemy C, Hamamah S, Lansac J. Cryopreservation of spermatozoa: a 1996 review. Hum Reprod Update. 1996;2(6):553-9. 14. Thachil JV, and Jewett, MA. Preservation techniques for human semen. Fertil Steril. 1981;35:546–8. 15. Holt, WV. Basic aspects of frozen storage of semen. Anim Reprod Sci. 2000; 62(1–3):3-22. 16. Sherman, JK. Cryopreservation of human semen. In: Handbook of the Laboratory Diagnosis and Treatment of Infertility. 1990; Keel B. and Webster BW, eds. Boca Raton, Fla, USA: CRC Press, pp. 229–59. 17. Fountain D, Ralston M, Higgins N, Gorlin, J, Uhl L, Wheeler C, et al. Liquid nitrogen freezers: a potential source of microbial contamination of hematopoietic stem cell components. Transfusion, 1997;37:585–91. 18. Abdelhafez F, Mohamed B, El-nashar S, Sabanegh E, Desai N. Techniques for cryopreservation of individual or small numbers of human spermatozoa : a systematic review. Hum Reprod. 2018;15(2):153–64. 19. Kuleshova LL, Lopata A. Vitrification can be more favorable than slow cooling. Fertil Steril. 2002;78(3):449-54. 20. Isachenko E, Isachenko V, Katkov II, Dessole S, Nawroth F. Vitrification of mammalian spermatozoa in the absence of cryoprotectants: from past practical difficulties to present success. Reprod Biomed Online. 2003;6(2):191-200. 21. Agha-Rahimi A, Khalili MA, Nabi A, Ashourzadeh S. Vitrification is not superior to rapid freezing of normozoospermic spermatozoa: effects on sperm parameters, DNA fragmentation and hyaluronan binding. Reprod Biomed Online. 2014;28(3):352-8. 22. Nawroth F, Isachenko V, Dessole S, Rahimi G, Farina M, Vargiu N et al. Vitrification of human spermatozoa without cryoprotectants. Cryo Letters. 2002;23:93–102. 23. Kamath, MS., Muthukumar, K., Appendix B: Solid Surface Vitrification. Methods Mol. Biol.2017;1568:297-307. 24. Rahiminia T, Hosseini A, Anvari M, Ghasemi-esmailabad S, Talebi AR. Modern human sperm freezing: Effect on DNA , chromatin and acrosome integrity. Taiwan J Obs Gynecol. 2017;56(Feb):472–6. 25. Said TM, Gaglani A, Agarwal A. Implication of apoptosis in sperm cryoinjury. Reprod Biomed Online. 2010;21(4):456-62. 26. Pugliesi G, Fürst R, Carvalho GR. Impact of using a fast-freezing technique and different thawing protocols on viability and fertility of frozen equine spermatozoa. Andrologia. 2014;46(9):1055-62. 27. Fürst R, Carvalho GR, Fürst MCO, Ruas JRM, Borges AM, et al. Efeito do resfriamento do sêmen eqüino sobre sua congelabilidade. Arq Bras Vet Zootec. 2005;57:599–607. 28. Shah SA, Andrabi SM, Qureshi IZ. Effect of equilibration times, freezing, and thawing rates on post-thaw quality of buffalo (Bubalus bubalis) bull spermatozoa. Andrology. 2016;4(5):972-6.b. 29. Vidament M, Yvon JM, Couty I, Arnaud G, Nguekam- Feugang J, et al. Advances in cryopreservation in modified INRA 82. Anim Reprod Sci 68:201–218. 30. Mazur P. Basic concepts in freezing cells. In: Proc. 1st International Conf. Deep Freezing Boar Semen. Uppsala, Sweden, 2005;91–111. 31. Amesse LS, Srivastava G, Uddin D, and Pfaff-Amesse T. Comparison of cryopreserved sperm in vaporous and liquid nitrogen. J Reprod Med. 2003;48:319–24. 32. Saritha KR, and Bongso, A. Comparative evaluation of fresh and washed human sperm cryopreserved in vapor and liquid phases of liquid nitrogen. J Androl. 2001;22:857–62. 33. Satirapod C, Treetampinich C, Weerakiet S, Wongkularb A, Rattanasiri S, et al. Comparison of cryopreserved human sperm from solid surface vitrification and standard vapor freezing method: on motility, morphology, vitality and DNA integrity. Andrologia. 2012;44(Suppl. 1):786–790. 34. Lim JJ, Shin TE, Song S, Bak CW, Yoon TK and Lee DR. Effect of liquid nitrogen vapor storage on the motility, viability, morphology, deoxyribonucleic acid integrity, and mitochondrial potential of frozen-thawed human spermatozoa. Fertil Steril. 2010;94:2736–41. 35. Gosalvez J, Nunez R, Fernandez JL, Lopez-Fernandez C, Caballero P. Dynamics of sperm DNA damage in fresh versus frozen–thawed and gradient processed ejaculates in human donors. Andrologia- 2011;43:373–377. 36. Isachenko E, Isachenko V, Katkov II, Rahimi G, Schondorf T, et al. DNA integrity and motility of human spermatozoa after standard slow freezing versus cryoprotectant-free vitrification. Hum. Reprod.2004;19:932–939. 37. Wu Y. Successful delivery derived from cryopreserved rare human spermatozoa with novel cryopiece. Am Soc Androl. 2017;5:832–7. 38. Gianaroli L et al. DNA integrity is maintained after freeze-drying of human spermatozoa. Fertil Steril. 2012;97(5):1067-73. 39. Ward MA, Kaneko T, Kusakabe H, Biggers JD, Whittingham DG and Yanagimachi R. Long-term preservation of mouse spermatozoa after freeze-drying and freezing without cryoprotection. Biol, 2003. 40. Arav A and Saragusty J. Directional freezing of sperm and associated derived technologies. Anim Reprod Sci; 2016, S0378-4320(16):30045-8. 41. Keskintepe L and Eroglu A. Freeze-Drying of Mammalian Sperm. In: Wolkers, W F and Oldenhof, H eds. Cryopreservation and Freeze-Drying Protocols. 3 ed. New York Heidelberg Dordrecht London: Springer Humana Press. 2015;.489-97. Authors: Iris Martínez Rodero and Raquel Pillado González "Multinucleated blastomeres (MNBs) present in embryos are morphological abnormalities of unclear origin, which have been extensively correlated with chromosomal defects, lower blastocyst formation and implantation rates". 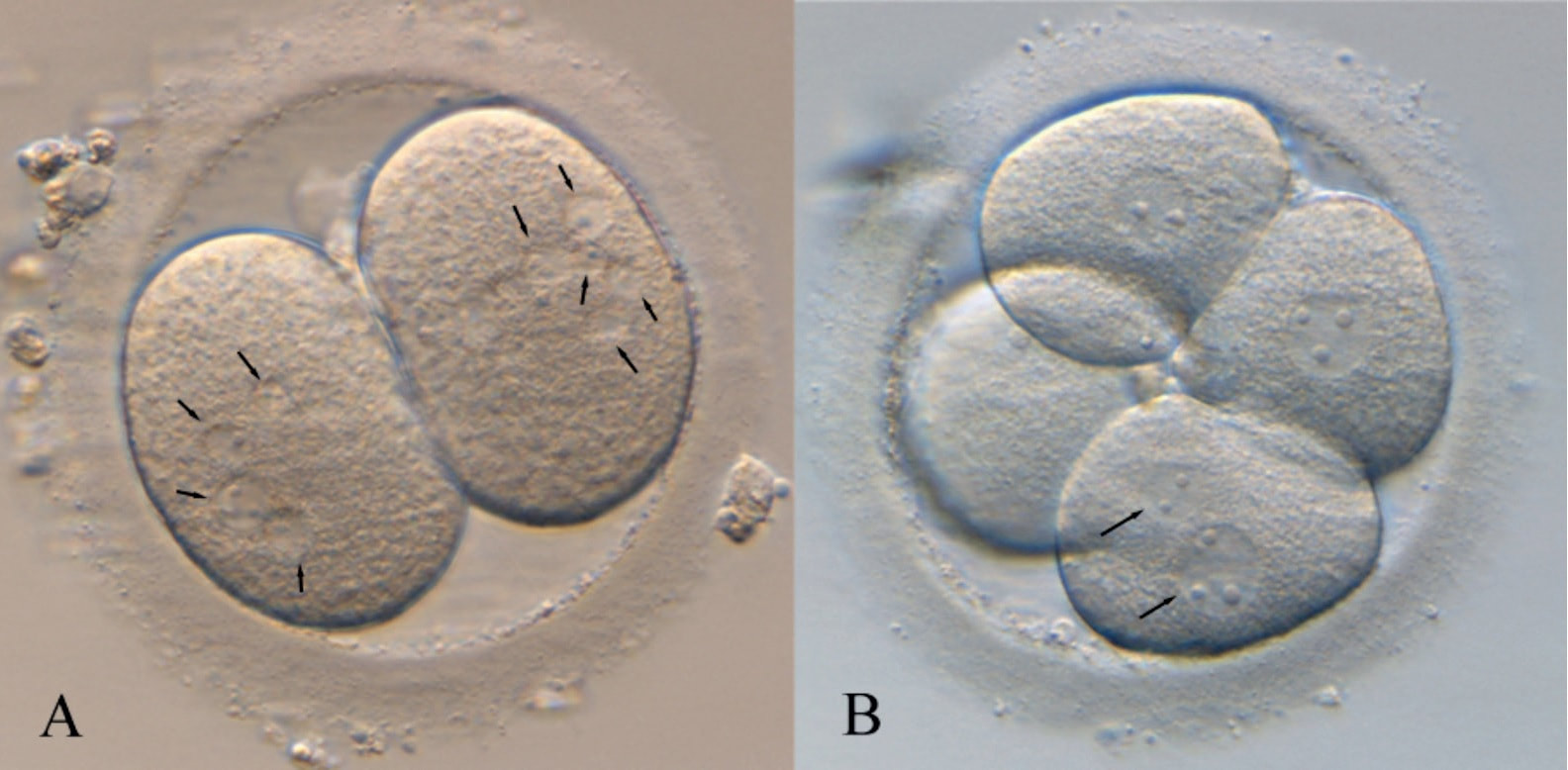 Fig 1. (A) Embryo displaying multi-/micronucleation in both blastomeres; (B) Embryo with binucleation in one of the blastomeres. Arrows point to the position of multiple nuclei (1). INTRODUCTION Selection of high-quality embryos is an important factor for the successful outcome of assisted reproduction technologies (ART). Nowadays, criteria for selection are mainly based on morphological features such as embryo fragmentation, cell number, blastomeres uniformity, etc. (2). The parameters studied so far have been demonstrated to be useful indicators of embryo quality. Their evaluation is performed through non-invasive light microscopy-based analyses, usually carried out once a day at specific time points. This approach intends to minimise the events of taking the embryos out of the incubators and exposing them to undesired harmful conditions. The presence of multinucleated blastomeres (MNBs) can be regarded as one of those indicators, and even though previous studies had already connected it to DNA abnormalities and low pregnancy rates as early as in the 90s (3,4,5) the origin of this phenomenon still remains unclear. Several possible factors seem to influence its genesis, but the specific cause for its occurrence is yet to be determined (6). Since the introduction of time-lapse imaging and monitoring technology, IVF laboratories have been able to carry out more exhaustive and continuous observations on embryo development, keeping risks at a minimum (7,8). By identifying the precise timing of specific key events of blastomere cell cycle interludes and of the embryo´s overall growth it is possible to assess its quality (7). Furthermore, time-lapse imaging and monitoring systems have facilitated the study of multinucleation (MN) in relation to its incidence in time and in the population, as well as its correlation with other morphological features and clinical variables (6,7,9). In the IVF context multinucleation is defined as the presence of two or more nuclei in one or more blastomeres. Multiples studies even differentiate between binucleated and multi-/micro-nucleated (three or more nuclei) blastomeres (Fig 1) (1,4,7). Reports on this phenomenon range from 17% to 69% of the total of cultured embryos, depending on the groups used within assays and authors (10). Factors that may influence MNB appearance are numerous and have been repeatedly studied (6,7,9,11). To date, several explanations have been proposed for MNBs: dysfunction of the mitotic spindle or the occurrence of karyokinesis without cytokinesis (12); DNA breaks or imperfect mitosis (13); nuclear membrane alterations (14) and even other factors not directly responsible for MNBs and yet linked to its presence (1). FACTORS THAT MAY RELATE TO THE APPEARANCE OF MULTINUCLEATION
Results from various studies have shown a difference in the percentage of multinucleated embryos between groups that had been fertilised by traditional IVF vs ICSI. Van Royen and colleagues showed 32.7% of MN embryo in the IVF group, compared to 34.5% in the ICSI group (6). Accordingly, Walmsley et al. 2003 reported 17,2% vs 18,3% of MN in embryos derived from IVF and ICSI, respectively (11).
Apparently, the type of infertility factor seems not to affect MN rates. Some studies have reported no significant differences in the percentages of MN embryos between cases of female factor-only infertility and male factor-only infertility (32.7% and 34.7%, respectively). In addition, differences were not found between these cases and those with both partners affected by some sort of infertility, either (6). However, different studies support the relevance of oocyte culture, specially regarding certain processes that occur naturally in in vivo conditions and that are essential for the proper embryo development. Data show that both oocytes subjected to negative conditions when cultured in vivo (like stern hypoxia, for instance) and oocytes cultured in vitro derive in a higher percentage of MN embryos (15,16). Regarding male infertility, results are more controversial; whereas some studies reflect a higher MN rate for cases in which male factor is especially severe (normally derived for ICSI rather than IVF) (11), other authors show no significant differences (6).
Records exhibit that cycles with an accelerated ovulation induction response present increased MN rates (6,17). Furthermore, several studies have reported that embryos derived from patients from whom ten or more oocytes had been collected presented a significantly higher MN rate than embryos from groups of nine or fewer oocytes (6,8,18). This is in accordance with the fact that patients who need high doses of FSH present higher MN rates (6). Very short cycles and cases where high doses of GnRH are needed trigger the development of high numbers of premature follicles that produce oocytes, which despite being able to reach metaphase II and become fertilized fail to go through proper nuclear cleavage (6,18). Lastly, even though differences in GnRH doses have been associated to significant differences in the incidence of MN embryos, similar results have not been observed when using different hormones (like rFSH, r-hFSH, purified urinary FSH or urinary gonadotropin, for instance) (6).
Multiple studies have been conducted on patients ranging from 25 to 45 years old. Several authors divided data in 5-year interval groups in order to verify whether patient age correlates with MN. However, the only significant difference was found when comparing women of +40 with younger ones of -35, presenting higher degree of MN in the first group (6,9).
Chromosome polymorphisms consist in heterochromatin variability. These are usually located in the long arms of chromosomes 1, 9 and 16, and the short arms of chromosomes from the groups D and G (13, 15 and 21, 22 and Y) (19). Even though such polymorphisms are generally regarded as normal within karyotypes (20), studies indicate that some of them might be associated with certain clinical problems such as abnormal spermatogenesis (21), infertility (22,23), recurrent miscarriages (24,25) and higher rate of chromosome abnormalities among blastomeres at the cleavage stage (26,27). Sun and collaborators hypothesized that couples with chromosome polymorphisms might experience a higher rate of embryo multinucleation (19). Nevertheless, the authors found no association between chromosome polymorphisms and MN embryo formation in couples undergoing IVF (19).
- Cellular fragmentation Although MN may appear regardless of the cellular fragmentation levels, several papers support the correlation between these two features (6,28,29). In particular, Van Royen et al. divided the level of fragmentation in three categories: F1 (≤10%), F2 (10-20%) and F3 (20-30%); this study presented evidence for higher MN in F2 and F3 when compared to F1, but similar to each other (6). - Cleavage rate When 3-cell and 5‐cell day-2 embryos were observed under the microscope, both types exhibited significantly higher multinucleation (28.2% -50%) than regular ones with the ideal 4-cell cleavage pattern (with only 16.8% MN). Similarly, day-3 embryos with the typical 8‐cell stage showed significantly lower multinucleation (15.5%) than 7‐cell and 9‐cell embryos (6). As it has been mentioned, ideal 4-cell and 8-cell stages show similar MN percentages. However, application of time-lapse imaging has revealed a significant decrease in MN from the 2-cell to the 4-cell stage (from 43.2% to 15.0%). The analysis of MN in 2-cell embryos indicated that, after cleavage, the majority (52%) of 2-cell MN embryos became mononucleated, whereas only a lower percentage (34%) showed MNBs, and about 14% were of poor quality (with only one or no visible nucleus at all) (9). This decrease in the MN rate suggests that 2-cell MN embryos are able to self-correct their nuclear abnormalities. But this repair mechanism has been observed in both euploid and aneuploid embryos, therefore it cannot be used as an indicator of chromosomal normality during embryo selection (8,9,30). An extended duration of both 2-cell and 4-cell stages has been proposed as a possible indicator of the occurrence of nuclear self-correction (9). INCIDENCE OF MULTINUCLEATION IN CLINICAL IVF As previously exposed, it is through time-lapse imaging that a far higher percentage of multinucleation (25%) has been detected compared to static observations on day 2 at 42 hours post-insemination (hpi) (<5%) (7). These observations have demonstrated that multinucleation is a frequent event that, according to Yilmaz et al., is present in at least one embryo in 41.3% of IVF cycles (31). Data provided by Desai and colleagues reported that approximately 56% of binucleated embryos and 48% of those with three or more nuclei went on to form blastocysts that met the appropriate criteria for vitrification (7). In addition, data from different studies point to binucleation being more frequent than blastomeres with 3 or more nuclei (7,31,32). At the same time, multinucleation has provided an additional criterion for embryo selection, since it is mainly observed in those of poor quality and is associated with direct and/or reverse cleavage (7). It has been observed that, out of all embryos found showing direct and/or reverse cleavage, at least one fourth were also multinucleated (7). By using time-lapse, multinucleation has been repeatedly observed to be a reversible event in a high proportion of embryos (7,32). Multinucleation reversibility has been reported to be as high as 73.4% (32); this has been calculated as the proportion of embryos in which multinucleation was detected at 2-cell stage, but not visible at 4-cell stage (likely due to self-correction mechanisms, as above-mentioned). In fact, Aguilar and collaborators reported 127 multinucleated embryos at 4-cell stage out of the 479 ones initially observed to present this feature at 2-cell stage. De novo multinucleation at the 4-cell stage in turn was observed in 36 embryos (32). IMPACT OF MULTINUCLEATION ON IVF OUTCOMES Multinucleation has traditionally been related to both low blastocyst formation (33) and implantation rates (5,6,17,28,34), and linked to the likely presence of chromosome abnormalities, which consequently results in embryo arrest (35). Nevertheless and despite all the existing evidences, there is still much controversy regarding multinucleation; reports have been published revealing cases in which fully binucleated 4-cell stage embryos had eventually developed into euploid blastocysts and genetically normal children (31,34).
Although some preimplantation genetic testing (PGT) studies have shown that not all multinucleated embryos are chromosomally abnormal (31,32,36) multinucleation is predominantly associated to chromosomal defects and poor implantation prognosis (3,31,37). Kligman and colleagues published that 74.5% of multinucleated embryos were chromosomally abnormal, compared to 32.3% of non-multinucleated embryos (3). Years later, Ambroggio et al. revealed an increased incidence of aneuploidy of MN 4-cell stage embryos when compared to single-nucleated embryos (85% vs 78%), suggesting that multinucleated embryos should not be recommended for transfer in IVF cycles (37). These results were confirmed when, from 395 MN embryos tested for PGT, Yilmaz et al. reported that 82.5% of MN blastomeres exhibited two nuclei, whereas the remaining blastomeres presented a single or three or more nuclei (31). Noteworthy, binucleated patterns of multinucleation may be less detrimental, since a high percentage of embryos with such feature are euploid, compared to embryos exhibiting three or more nuclei in a single blastomere (38).
Embryo morphokinetics was studied and related to the multinucleation status in a study conducted by Meseguer’s team (32). In the study, 53.4% of a total 1676 embryos included were MN. Based upon the reported data, differences in morphokinetics between multinucleated and non-multinucleated embryos at both 2-cell and 4-cell stages comprise cleavage events involving the completion of the first mitosis and the length of the S-phase. These differences affected the following parameters: t2, t3, second cell cycle (cc2=t3-t2), t4, t6, t7 and t8. These results allowed to conclude that, if multinucleation remains at 4-cell stage, it takes longer for the embryos to complete the next cell cycle (cc3=t5-t3). Should this be true, the restoration system would not be efficient if MNBs were still observed after the 2-cell stage (32). The origin of the multinucleation phenotype has been suggested to be multiple: disruption of intracellular restructuring, remodelling or imprinting in the developing oocyte, or even alterations in DNA replication, cytokinesis or compaction during the first cell cycle (16). If multinucleation appears as a result of defects in cell function, differences in morphokinetics between MN and non-MN embryos during these early stages may be expected (32).
Opinions on the impact of the multinucleation phenotype on implantation rates diverge from each other: On one hand, cell stage for MN appearance has been proposed to exert the highest effect on the implantation rate. Authors supporting this claim are divided into two positions: those who affirm that the presence of MN at the 2-cell stage is actually insignificant in terms of differences on implantation rates, but it is at 4-cell stage when it does have a measurable negative effect (32); and the authors who argue implantation rates to be significantly reduced when MN is already observed at the 2-cell stage (8). On the other hand, the school of Meriano and coauthors affirm that binucleation is less harmful than any other type of multinucleation (16). However, Aguilar and colleagues explained that their differences with Meriano were found on the frequency of image acquisition and the systems used to measure multinucleation (32); whereas the former acquired one picture in seven different focal planes every 20 minutes, the latter recorded images every 2.5 minutes (32). In any case, it has been demonstrated that patterns of multinucleation at 4-cell stage are correlated with low implantation rates, while any of the other cases has been reported to decrease the chances to achieve pregnancy (16,32). CONCLUSIONS Multinucleation is a common and reversible event observed in human IVF embryos, and it is specially frequent as binucleation at two-cell stage. It is associated with chromosomal defects and altered morphokinetic parameters, eventhough binucleation patterns seem to be the less severe. Regarding multinucleation impact on implantation rate, results are controverted. It seems that implantation rates are not affected when multinucleation appears as two nuclei in two-cell stage. Although the presence of multinucleated blastomeres in human embryos has been associated with the above-mentioned undesired characteristics in IVF embryos, the reasons explaining its appearance and occurrence in time and its relationship with patient specifications have not been deeply studied until time-lapse systems became available. Even though different causes have been suspected to lie behind MNB development, none of them have been proved actually represent the main responsible. Nevertheless, a growing number of studies provide data untangling the relationship between MN and assisted reproduction fertilization methods (IVF and ICSI), stimulation cycles, infertility factors, culture conditions and other embryonic morphological characteristics. Even though sometimes results from different studies may seem contradictory, this might be accredited to the differences in sample sizes. All the above said, it seems reasonable to highlight the need for further research on this issue. It would be highly helpful to unveil the actual triggers of multinucleation, to develop optimal ART practices that avoid increasing MN incidence, and to unravel any other correlation with adverse embryo features during development. Deeper knowledge would help improving embryo assessment methods and, consequently, increase the rates of successful ART outcomes. REFERENCES
Authors: María Caballero Sastre & Lidón Carretero Vilarroig “Due to the current lifestyle and habits, high production of ROS and oxidative stress are the most common mechanisms responsible for DNA damage”  Fig 1. Example of two techniques to detect DNA fragmentation in a sperm sample. A) TUNEL assay: sperm in green indicate DNA fragmentation. B) Sperm Chromatin Dispersion test: the halo around both central sperm nuclei indicate non-fragmented DNA, while the absence of halo in the sperm in the upper right hand corner indicates DNA fragmentation (1). INTRODUCTION Male factor plays a role in 33% of couples experiencing infertility problems. Although it is less understood than its female counterpart, a great effort has been made in order to understand its basis. The World Health Organization (WHO) periodically updates its Laboratory Manual for the Examination and Processing of Human Semen, as well as the reference parameters (2). However, since results from conventional seminograms vary depending on temporal factors such as stress, period of abstinence or sample collection, it is difficult to diagnose male infertility using conventional semen analyses (3). This makes also necessary to understand the process behind male gamete formation (spermatogenesis) in order to achieve a better comprehension of the molecular basis of male infertility. Within spermatogenesis, meiotic problems may occur resulting in sperm cells carrying chromosome alterations, and during spermiogenesis sperm DNA becomes highly compacted by replacing histones by protamines. Humans express two protamines (P1 and P2) in a 1:1 ratio; an imbalance of this ratio results in male infertility (2). Measurement of sperm DNA integrity as a marker of sperm quality has recently gained interest for embryologists. The study of sperm DNA fragmentation is highly indicated in cases of implantation failure or repeated miscarriages. There is a wide range of tests currently used aimed to analyze sperm DNA integrity (Fig. 1). These techniques make it possible to determine whether there is a high rate of single-strand or double-strand breakpoints (SSB or DSB) in the DNA; in such cases the fertilizing and replicative ability of the sperm cell may be jeopardised. CONVENTIONAL SEMEN ANALYSIS Conventional semen analyses are the first step to measure semen parameters. Using fresh ejaculated semen, andrologists evaluate sperm motility, vitality and morphology, as well as its concentration along with other seminal components, and total volume of the ejaculate. WHO gives the following results as a reference for normality (2): - Volume of at least 1.5 mL. The total amount of fresh ejaculated semen may vary depending on the abstinence period. Thus, a period of abstinence of 3-7 days before the analysis is recommended. - Concentration of at least 15 million sperm cells per millilitre (15 mil/mL). Samples with less than 15 mil/mL are classified as oligozoospermic, whereas samples with no sperm cells are classified as azoospermic. Samples with a concentration below 100,000 sperm cells per millilitre are considered cryptozoospermic. - Motility and vitality. Even though sperm cells acquire their characteristic forward and rapid movement during capacitation, the way they move within a fresh sample is a quality indicator. Proper movement consists in progressive and linear displacement, while those sperm cells with circular or even missing movement are considered less capable of fertilising. Such samples are classified as asthenozoospermic. Following these WHO’s reference guidelines, sperm samples are considered normal if linear progressive movement is found in at least 32% of the cells. - Correct morphology in at least 4% of the sperm cells. This feature refers to (4): (i) sperm cells with one head and one tail; (ii) sperm cells with a round/oval-shaped head; (iii) lack of cytoplasmic droplet. Samples with less than 4% of morphologically normal sperm cells are classified as teratozoospermic. Fluorescent In Situ Hybridization (FISH) in spermatozoa is a complementary technique to conventional semen analyses. This test is indicated in cases of altered seminal values, miscarriages, implantation failure, non-oocyte-driven fertilization failure cause or anomalous embryonic development (5). The objective of this procedure is to verify the chromosome number in human sperm cells. As haploid cells, these must contain 23 chromosomes each. FISH assays use fluorescent probes to recognize specific regions of certain chromosomes in every test. The 5-probe FISH test analyzes chromosomes X, Y, 13, 18 and 21, while the 9-probe FISH test also includes chromosomes 15, 16, 17 and 22. Altered results can suppose a high percentage of diploid, dysomic or hyperploidic sperm cells, which may compromise fertilization and embryo development (Fig. 2). In such cases, a cycle of in vitro fertilization is indicated, followed by preimplantation genetic test (IVF-PGT) by aneuploidy screening, in order to transfer only euploid embryos. Due to all different variables that may influence the result of a conventional semen analysis (6-8) and FISH assay (7), DNA fragmentation analysis has gained attention over the last years.  Fig 2. FISH analysis of testicular human sperm. Sperm nuclei hybridized by chromosome 18 (green) and Y (red) probes. (a) Diploid sperm nucleus exhibiting signals for chromosomes 18,18 and YY.. (b) Nucleus with an absent signal for chromosome 18 (left) and nucleus displaying gonosomal dysomy for the Y chromosome (5). SPERM DNA FRAGMENTATION According to Sakkas et al., DNA damage in spermatozoa can be induced by six main mechanisms: [1] apoptosis during spermatogenesis, [2] DNA strand breaks produced during remodeling of sperm chromatin typical of spermiogenesis, [3] oxygen radicals during sperm transport through the seminiferous tubules and the epididymis, [4] endogenous caspases and endonucleases, [5] radiotherapy and chemotherapy and [6] environmental toxicants (8). Some of these mechanisms seem to be involved in specific cases of DNA damage due to a variety of factors such as varicocele (10), bacterial infections (11), age (15) or abstinence (13). The main types of DNA damage include are mismatch, base adducts, crosslink, base modification, pyrimidine dimerization, abasic sites, thymidine glycol and oxidation at guanosine sites. When evaluating DNA fragmentation, two kind of breaks are considered: single strand breaks (SSBs) and double strand breaks (DSBs) (Fig. 3). Although both type of DNA lesions are present in sperm of both fertile and infertile men, the latter have been observed to present higher levels of DNA fragmentation (14). 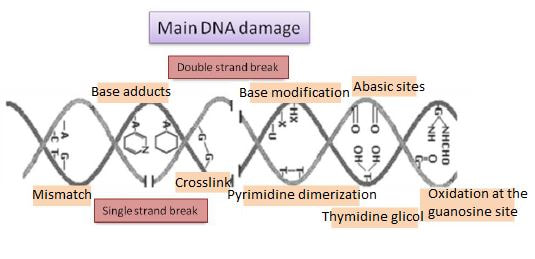 Fig 3. Main types of lesions found to be induced in the DNA (14). Each kind of DNA break has a different origin. For instance, nucleases usually generate both types, whereas chromatin remodelling may cause DSBs produced by topoisomerase II and reactive oxygen species (ROS), and other radical molecules generate mainly SSBs, DNA adducts and abasic sites (16, 17). Whereas SSBs are slightly detrimental and easy to repair, DSBs represent serious harmful lesions that can lead to chromosome aberrations (16, 18). Fortunately, cells present different pathways to face and repair DNA damage. The main mechanisms present in germline cells are direct reversal repair, nucleotide excision repair, base excision repair, mismatch repair, post replication repair and DNA double strand break repair (19). These DNA repair processes can take place during spermiogenesis (20), in the fertilized oocyte (21) or during early embryonic development (22). The capacity to repair DNA damage depends on the type of DNA damage present in the sperm; should it fail, the embryo may not develop or miscarriage may eventually occur (14). DIFFERENT TECHNIQUES TO ASSES DNA DAMAGE - Sperm Chromatin Structure Assay (SCSA): This technique, first proposed in 1980 by Evenson and coauthors (23), uses flow cytometry to measure sperm DNA integrity. It tests the vulnerability of DNA to acid-induced denaturation by exposure to acridine orange media (AO) (24). AO binds to both double-stranded and single-stranded DNA, but fluoresces in different colour, being red or green, respectively. The DNA fragmentation index (DFI), which represents the percentage of spermatozoa with red fluorescence, is analyzed and represented on a histogram. Green fluorescence in turn shows the percentage of mature undamaged sperm (24). - TUNEL test: This test detects SSBs and DSBs by labelling 3’-free ends of DNA fragments (25) with fluorescent dUTP nucleotides in a reaction catalyzed by the terminal deoxynucleotidyl transferase. The evaluation process can be performed using light and fluorescence microscope or flow cytometry (26, 27). - Single cell gel electrophoresis assay (COMET): It enables the detection of both SSB and DSBs. It consists in the lysis of sperm cells with detergents and high salt-density components. After lysis, the sample is introduced into a gel and subjected to an electrophoretic field resulting in the migration of both DNA and proteins. DNA fragments move faster than intact DNA; as a consequence, fragmented DNA from sperm cells migrate through the gel leaving a comet-like trace, whereas DNA without breaks is seen as a dot. Samples can be analyzed by fluorescence microscopy. The ratio between comet-like trace and dots defines the degree of fragmentation of the sample.  Fig 4. Image analysis on different human sperm groups. (A) Control sperm DNA; (B) sperm sample with no DNA damage; (C) sperm sample with low rate of DNA fragmentation; (D) sperm cells with high level of DNA damage showing a comet-like trace (28). - Sperm cromatin dispersion (SCD) test: SCD test allows for the differentiation between sperm with fragmented DNA and intact DNA. This technique denatures DNA that contains breaks using an acid solution. Subsequently, spermatozoa are treated with a lysis buffer to remove membranes and proteins and, in so doing, a peripheral halo of DNA loops is formed around the sperm. The observation of a large halo means the sperm nucleus presents low levels of DNA fragmentation. On the contrary, a small or absent halo is indicative of highly fragmented DNA (Fig. 5). Results can be evaluated using either fluorescence or bright-field microscopy (29).  Fig 5. Results of SCD test with bright-field microscopy after Wright staining. Sperm with no halo contain fragmented DNA (asterisk) (29). - γH2AX test: Not only is γH2AX labelling assay used to assess DNA fragmentation in sperm cells, but it has been also used in order to evaluate DNA damage in tumour cells during radio- or chemotherapy, thus providing a potential prediction marker (30). Once DSBs occur, histone H2AX becomes phosphorylated at Ser139 (γH2AX), playing a role in signalling the break sites to be repaired (31). The presence of γH2AX in this context is commonly assessed by flow cytometry, being the percentage of γH2AX higher among infertile men. LIFESTYLE AND POTENTIAL HARMFUL FACTORS FOR DNA Among the six mechanisms above mentioned that can produce DNA damage, oxidative stress and ROS are the most common (17), which is mainly due to current lifestyle and habits. The term ROS includes highly oxidative radicals like OH, non-radical species like O2- and H2O2, as well as reactive nitrogen species. Spermatozoa produce ROS themselves, since these are required for several functions such as sperm motility, capacitation, acrosomal reaction, zona pellucida binding (32) and chromatin compaction (17). Because ROS concentration can be harmful, it is important to guarantee the action of antioxidant mechanisms in order to maintain cell homeostasis (33). Therefore, decompensation in ROS final concentration, due to either excess production or to failure in antioxidant mechanisms, would eventually lead to oxidative stress and, as a consequence, to DNA damage (34-36). Alcohol and smoking Several studies have demonstrated both alcohol and tobacco to be responsible for the significant increase in ROS at sperm production (37,38). Although some studies have shown reduced efficiency of antioxidant mechanisms in smokers and people with a high intake of alcohol (39), contradictory results have been reported as well (40). These studies showed an unbalance in favour of ROS despite the increased antioxidant activity, suggesting the antioxidant mechanisms were somehow unable to protect from oxidative damage (40). The various chemical products found in tobacco such as cadmium, lead (17) and nicotine, and alcohol itself most likely seem to result harmful for semen parameters, since they have been reported to cause DNA damage (40). Obesity A recent study by Jin-Chun Lu and collaborators has shown data from a correlation between sperm DNA fragmentation index and obesity (41). Samples from 1010 subfertile men from 18 to 50 years old were analysed using SCSA. Although the authors found no correlation between obesity and sperm index fragmentation, they did find a positive correlation between sperm fragmentation and seminal plasma triglyceride and total cholesterol levels.A separate study on these matters concluded there is no correlation between body mass index and DNA integrity. The authors stated obesity might affect lipid metabolism and reproductive hormones, leading to a decreased sperm quality (42). Varicocele Varicocele is an enlargement of the veins located within the scrotum. It is postulated that varicocele causes a rise in temperature that leads to higher DNA fragmentation ratio and ROS production (43). About 15% of adult men suffer from this condition (44), but authors suggest that varicocelectomy can improve semen parameters after 3 months (8), which is the expected period for de novo sperm production in humans. Environmental contaminants Exposure to environmental contaminants has been demonstrated to be the main cause for male infertility, even over genetic defects (45). These substances, known as endocrine disruptors (ED) (Table 1), can negatively affect the endocrine system by mimicking natural hormones in the organism like estrogens and androgens, and so they can block interactions between natural hormones and their receptors (46). Exposure of an individual to ED may cause important specific epigenetic modifications in sperm (that are eventually passed down to several generations), as well as severe fertility disorders (45). 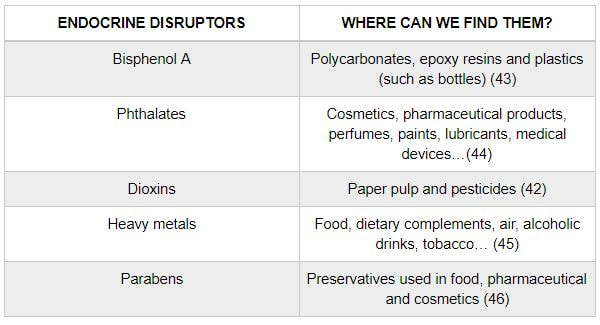 Table 1. EDs and common localisation. Electromagnetic radiation: mobile phones radiation and Wi-Fi It has been suggested that the electromagnetic radiation (EMR) emitted by some electrical devices such as mobile phones or Wi-Fi suppresses the natural antioxidant mechanisms and increase the oxidative stress, which consequently increases infertility (50). Although studies in both animals and humans have demonstrated the detrimental effect of EMR, there is still controversy. Some studies show that the prolonged use of mobile phones decreases sperm motility (51), concentration (52), viability and morphology (53), while others have found no effects on sperm concentration (reviewed in 54). Moreover, results have been reported on the increase in DNA fragmentation in human sperm associated to exposure to Wi-Fi networks (51). Heat It is important to maintain testicular temperature 2-4ºC lower than the body temperature for spermatogenesis to progress normally. This justifies the existence of several thermoregulatory mechanisms found in the organism, such as the characteristics of the scrotal sac, the musculature and the vasculature (18). Fertility is directly affected by a rise in temperature due to the increased DNA fragmentation index (55) and sperm cell apoptosis (18). Several factors can cause hyperthermia, namely: the use of tight clothes, saunas, laptops and mobile devices closely located to the testes (18), or even an episode of high fever or varicocele (55). Moreover, the severity of damage varies with duration, frequency and intensity of heat (18). ANTIOXIDANT THERAPY Spermatozoa present low levels of antioxidants and DNA repair enzymes. Therefore, the presence of these factors in the seminal plasma is critical. As it has been demonstrated, not only subfertile men have been observed to present lower levels of antioxidants than fertile men (35, 56), but they have been also found to exhibit higher levels of ROS (36). Antioxidants present in semen include vitamins C and E, zinc, selenium, carnitine and carotenoids (57). These components can be found in the food (table 2), meaning that controlling the dietary antioxidant intake, oxidative stress can be minimised or avoided and, as a consequence, DNA damage, too (17). Besides the above-mentioned antioxidants, polyunsaturated fatty acids are also an important source of antioxidants. They are classified into omega-3, omega-6 and omega-9 fatty acids and, with the exception of omega-9, they need to be incorporated into the diet through the intake of vegetable and fish oils. 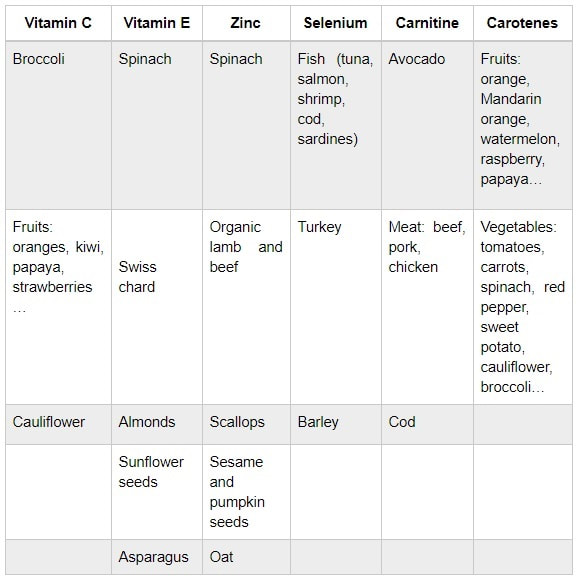 Table 2. Antioxidant-rich food. CONCLUSIONS The first step to check male infertility in a couple undergoing infertility issues is the completion of a conventional semen analysis established by the WHO. This kind of test evaluates several parameters such as motility, vitality, morphology, concentration and total volume of the ejaculate. Because of the evidenced involvement of sperm DNA damage in infertility, its study has been gaining significance and, consequently, it is usually indicated in certain cases such as implantation failure or recurrent miscarriages. Techniques like SCSA, SCD, TUNEL, COMET and γH2AX test are normally used to analyze sperm DNA integrity. These approaches are helpful to discriminate between SSBs and DSBs or other types of DNA damage. Due to the current lifestyle and habits, high production of ROS and oxidative stress are the most common mechanisms responsible for DNA damage. Among such habits and lifestyle it is worth mentioning alcohol and smoking, environmental contaminants, electromagnetic radiations and heat sources. An efficient method to prevent DNA fragmentation is to resort to the intake of specific products as an antioxidant dietary therapy. Thus, a proper diet rich in such components may be able to reduce the incidence of sperm DNA fragmentation, and, consequently, to also improve male fertility. REFERENCES
1. Schulte RT, Ohl DA, Sigman M, Smith GD. Sperm DNA damage in male infertility: Etiologies, assays, and outcomes. J Assist Reprod Genet. 2010;27(1):3–12. 2. Edition F. Examination and processing of human semen. World Health. 2010;Edition, V(10):286. 3. Saleh RA, Agarwal A, Nelson DR, Nada EA, El-Tonsy MH, Alvarez JG, et al. Increased sperm nuclear DNA damage in normozoospermic infertile men: A prospective study. Fertil Steril. 2002;78(2):313–8. 4. Menkveld R. Clinical significance of the low normal sperm morphology value as proposed in the fifth edition of the WHO Laboratory Manual for the Examination and Processing of Human Semen. Asian Journal of Andrology. 2010;12(1):47-58. 5. J. Bataller Sánchez, A. Barberá Alberola, X. Vendrell Montón, M. Ruiz Jorro CCL. Valoración del área media de la cabeza espermática como posible indicador para solicitar FISH en espermatozoides. ASEBIR 2017. 6. Dunphy BC, Neal LM, Cooke ID. The clinical value of conventional semen analysis. Fertil Steril. Elsevier Masson SAS; 1989;51(2):324–9. 7. Intasqui P, Camargo M, Antoniassi M, Barradas V, Spaine DM, Bertolla RP. Sperm functional testing and conventional semen analysis: correlations, distributions, and putative mechanisms. Fertil Steril. Elsevier Ltd; 2013;100(3):S214. 8. Kruger T. Critical appraisal of conventional semen analysis in the context of varicocele. Asian J Androl. 2016;18(2):202. 9. Sakkas D, Alvarez JG. Sperm DNA fragmentation: mechanisms of origin, impact on reproductive outcome, and analysis. Fertil Steril. 2010;93(4):1027–36. 10. Roque M, Esteves SC. Effect of varicocele repair on sperm DNA fragmentation: a review. Int Urol Nephrol. Springer Netherlands; 2018;50(4):583–603. 11. González-Marín C, Roy R, López-Fernández C, Diez B, Carabaño MJ, Fernández JL, et al. Bacteria in bovine semen can increase sperm DNA fragmentation rates: A kinetic experimental approach. Anim Reprod Sci. 2011;123(3–4):139–48. 12. Vagnini L, Baruffi RLR, Mauri AL, Petersen CG, Massaro FC, Pontes A, et al. The effects of male age on sperm DNA damage in an infertile population. Reprod Biomed Online. 2007;15(5):514–9. 13. Comar VA, Petersen CG, Mauri AL, Mattila M, Vagnini LD, Renzi A, et al. Influence of the abstinence period on human sperm quality: Analysis of 2,458 semen samples. J Bras Reprod Assist. 2017;21(4):306–12. 14. González-Marín C, Gosálvez J, Roy R. Types, causes, detection and repair of DNA fragmentation in animal and human sperm cells. Int J Mol Sci. 2012;13(11):14026–52. 15. Vagnini L, Baruffi RLR, Mauri AL, Petersen CG, Massaro FC, Pontes A, et al. The effects of male age on sperm DNA damage in an infertile population. Reprod Biomed Online. 2007;15(5):514–9. 16. Cortés-Gutiérrez EI, López-Fernández C, Fernández JL, Dávila-Rodríguez MI, Johnston SD, Gosálvez J. Interpreting sperm DNA damage in a diverse range of mammalian sperm by means of the two-tailed comet assay. Front Genet. 2014;5:1–11. 17. Wright C, Milne S, Leeson H. Sperm DNA damage caused by oxidative stress: Modifiable clinical, lifestyle and nutritional factors in male infertility. Reprod Biomed Online. Reproductive Healthcare Ltd.; 2014;28(6):684–703. 18. Durairajanayagam D, Agarwal A, Ong C. Causes, effects and molecular mechanisms of testicular heat stress. Reprod Biomed Online. Reproductive Healthcare Ltd.; 2015;30(1):14–27. 19. Jaroudi S, Kakourou G, Cawood S, Doshi A, Ranieri DM, Serhal P, et al. Expression profiling of DNA repair genes in human oocytes and blastocysts using microarrays. Hum Reprod. 2009;24(10):2649–55. 20. Baarends WM, van der Laan R, Grootegoed JA. DNA repair mechanisms and gametogenesis. Reproduction. 2001;121(1):31–9. 21. Men NT, Kikuchi K, Furusawa T, Dang-Nguyen TQ, Nakai M, Fukuda A, et al. Expression of DNA repair genes in porcine oocytes before and after fertilization by ICSI using freeze-dried sperm. Anim Sci J. 2016;87(11):1325–33. 22. Derijck A, Van der heijden G, Giele M, Philippens M, De boer P. DNA double-strand break repair in parental chromatin of mouse zygotes, the first cell cycle as an origin of de novo mutation. Hum Mol Genet. 2008;17(13):1922–37. 23. Evenson DP, Darzynkiewicz Z MM. Relation of mammalian sperm chromatin heterogeneity to fertility. Science. 1980;210(4474):1131–3. 24. Khatun A, Rahman MS, Pang M-G. Clinical assessment of the male fertility. Obstet Gynecol Sci. 2018;61(2):179–91. 25. Ribas-Maynou J, García-Peiró A, Fernández-Encinas A, Abad C, Amengual MJ, Prada E, et al. Comprehensive analysis of sperm DNA fragmentation by five different assays: TUNEL assay, SCSA, SCD test and alkaline and neutral Comet assay. Andrology. 2013;1(5):715–22. 26. Rex AS, Aagaard J, Fedder J. DNA fragmentation in spermatozoa: a historical review. Andrology. 2017;5(4):622–30. 27. Kucuk N. Sperm DNA and detection of DNA fragmentations in sperm. Türk Üroloji Dergisi/Turkish J Urol [Internet]. 2018;44(1):1–5. 28. Pandir D. Assessment of the DNA Damage in Human Sperm and Lymphocytes Exposed to the Carcinogen Food Contaminant Furan with Comet Assay. 2015;58:773–80. 29. Fernández JL, Muriel L, Goyanes V, Segrelles E, Gosálvez J, Enciso M, et al. Simple determination of human sperm DNA fragmentation with an improved sperm chromatin dispersion test. Fertil Steril. 2005;84(4):833–42. 30. Xuan Huang H. Dorota Halicka Frank Traganos Toshiki Tanaka Akira Kurose Zbigniew Darzynkiewicz. Cytometric assessment of DNA damage in relation to cell cycle phase and apoptosis. Cell Prolif. 2005;8(4):223-43. 31. Jha KN, Tripurani SK, Johnson GR. TSSK6 is required for γH2AX formation and the histone-to-protamine transition during spermiogenesis. J Cell Sci. 2017;130:1835–44. 32. Doshi S, Khullar K, Sharma R, Agarwal A. Role of reactive nitrogen species in male infertility. Reprod Biol Endocrinol. 2012;10:109. 33. Sharma RK1 AA. Role of reactive oxygen species in male infertility. Urology. 1996;48(6):835–50. 34. Muratori M, Tamburrino L, Marchiani S, Cambi M, Olivito B, Azzari C, et al. Investigation on the Origin of Sperm DNA Fragmentation: Role of Apoptosis, Immaturity and Oxidative Stress. Mol Med. 2015;21(1):109–22. 35. R2 BSD. Oxidative stress: Major executioner in disease pathology, role in sperm DNA damage and preventive strategies. Front Biosci. 2017;9:420–47. 36. Aktan G, Doǧru-Abbasoǧlu S, Küçükgergin C, Kadioǧlu A, Özdemirler-Erata G, Koçak-Toker N. Mystery of idiopathic male infertility: Is oxidative stress an actual risk? Fertil Steril. 2013;99(5):1211–5. 37. Tweed JO, Hsia SH, Lutfy K, Friedman TC. The endocrine effects of nicotine and cigarette smoke. Trends Endocrinol Metab. Elsevier Ltd; 2012;23(7):334–42. 38. Anderson RA Jr, Willis BR, Oswald C, Reddy JM, Beyler SA ZL. Hormonal imbalance and alterations in testicular morphology induced by chronic ingestion of ethanol. Biochem Pharmacol. 1980;29(10):1409–19. 39. Lesgards JF, Durand P, Lassarre M, Stocker P, Lesgards G, Lanteaume A, et al. Assessment of lifestyle effects on the overall antioxidant capacity of healthy subjects. Environ Health Perspect. 2002;110(5):479–86. 40. Aboulmaouahib S, Madkour A, Kaarouch I, Sefrioui O, Saadani B, Copin H, et al. Impact of alcohol and cigarette smoking consumption in male fertility potential: Looks at lipid peroxidation, enzymatic antioxidant activities and sperm DNA damage. Andrologia. 2018;50(3):1–7. 41. Lu JC, Jing J, Chen L, Ge YF, Feng RX, Liang YJ, et al. Analysis of human sperm DNA fragmentation index (DFI) related factors: A report of 1010 subfertile men in China. Reprod Biol Endocrinol. Reproductive Biology and Endocrinology; 2018;16(1):1–9. 42. Bandel I, Bungum M, Richtoff J, Malm J, Axelsson J, Pedersen HS, et al. No association between body mass index and sperm DNA integrity. Hum Reprod. 2015;30(7):1704–13. 43. Silber S. The varicocele argument resurfaces. J Assist Reprod Genet. Journal of Assisted Reproduction and Genetics; 2018 44. Lundy SD, Sabanegh ES. Varicocele management for infertility and pain: A systematic review. Arab J Urol. 2017;16(1): 157-170 45. Jenardhanan P, Panneerselvam M, Mathur PP. Effect of environmental contaminants on spermatogenesis. Semin Cell Dev Biol. Elsevier Ltd; 2016;59:126–40. 46. Claude Monneret. What is an endocrine disruptor? C R Biol. Academie des sciences; 2017;340:403–5. 47. Fromme H, Bolte G, Koch HM, Angerer J, Boehmer S, Drexler H, et al. Occurrence and daily variation of phthalate metabolites in the urine of an adult population. Int J Hyg Environ Health. 2007;210(1):21–33. 48. Wirth JJ, Mijal RS. Adverse effects of low level heavy metal exposure on male reproductive function. Syst Biol Reprod Med. 2010;56(2):147–67. 49. Castelain F, Castelain M. Parabens: A real hazard or a scare story? Eur J Dermatology. 2012;22(6):723–727. 50. La Vignera S, Condorelli RA, Vicari E, D’Agata R, Calogero AE. Effects of the exposure to mobile phones on male reproduction: A review of the literature. J Androl. 2012;33(3):350–6. 51. Choy JT, Brannigan RE. Re: Use of laptop computers connected to internet through Wi-Fi decreases human sperm motility and increases sperm DNA fragmentation. Eur Urol. 2012;62(6):1196–7. 52. Kesari KK, Behari J. Microwave exposure affecting reproductive system in male rats. Appl Biochem Biotechnol. 2010;162(2):416–28. 53. Agarwal A, Deepinder F, Sharma RK, Ranga G, Li J. Effect of cell phone usage on semen analysis in men attending infertility clinic: an observational study. Fertil Steril. 2008;89(1):124–8. 54. Adams JA, Galloway TS, Mondal D, Esteves SC, Mathews F. Effect of mobile telephones on sperm quality: A systematic review and meta-analysis. Environ Int. Elsevier B.V.; 2014;70:106–12. 55. Ahmad G, Moinard N, Esquerr-Lamare C, Mieusset R, Bujan L. Mild induced testicular and epididymal hyperthermia alters sperm chromatin integrity in men. Fertil Steril. 2012;97(3):546–53. 56. Tremellen K. Oxidative stress and male infertility — a clinical perspective. 2008;14(3):243–58. 57. Talevi R, Barbato V, Fiorentino I, Braun S, Longobardi S, Gualtieri R. Protective effects of in vitro treatment with zinc, d-aspartate and coenzyme q10 on human sperm motility, lipid peroxidation and DNA fragmentation. Reprod Biol Endocrinol. 2013;11(1):1–10. Authors: Iris Martínez Rodero and Raquel Pillado González "Selection of the best spermatozoon for ICSI is critical and it is directly linked to fertilization rates, optimal embryo development and implantation rates" 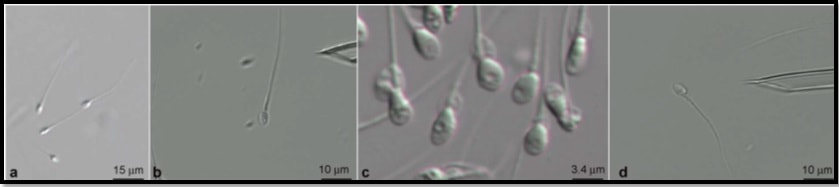 Figure 1. Spermatozoa observed at low magnification (a) and high magnification (b–d). The shape and presence of vacuoles can be clearly observed in (c). A vacuole-free spermatozoon is shown in (d). INTRODUCTION It is estimated that approximately 10% of couples seek help to overcome infertility problems. Data from the American Society of Reproductive Medicine points to male factor as the reason behind one third of couples’ infertility problems (1). The remaining two thirds seem to be equally distributed between female-related problems and a mixture of unexplained causes and combinations of male and female factors, respectively (1). As continuation of our previous post on sperm selection, we revisit the topic from the perspective of the numerous techniques currently available to be specifically used prior intracytoplasmic sperm injection (ICSI). This procedure is the advised treatment for most cases of male factor infertility; in fact, ICSI accounts for 70-80% of cycles performed globally (2). ICSI candidates often present low sperm count and/or increased probability of genetic abnormalities, compromised DNA and morphological defects of spermatozoa. Therefore, selection of the best spermatozoon for ICSI is critical and it is directly linked to fertilization rates, optimal embryo development and implantation rates (3). Traditional sperm selection procedures (swim-up and density gradient centrifugation (DGC)) are based just on motility rather than molecular features. Consequently, in order to choose the best spermatozoa, researchers have developed different non-invasive methods to process and select sperm for ICSI (Table 1) (2). 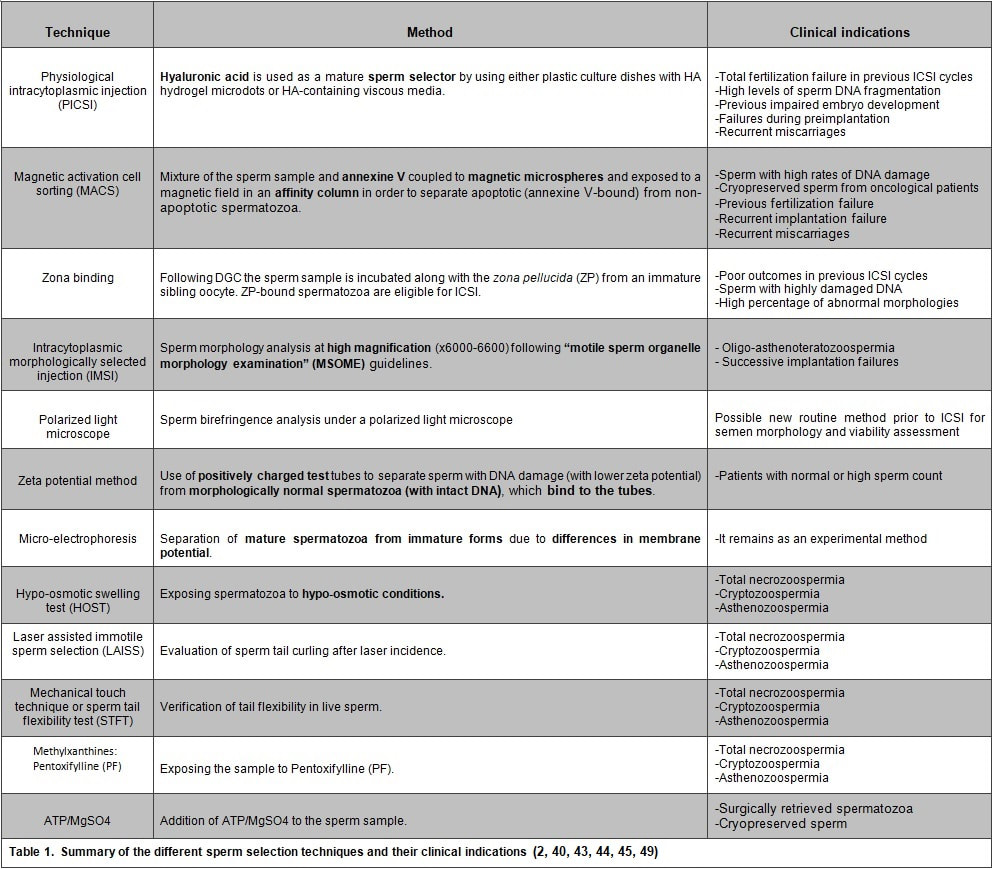 SPERM SELECTION TECHNIQUES Sperm selection prior to fertilization is a crucial step for IVF success, especially in cases of low number of available oocytes to inject (4). Several techniques are currently utilised for this purpose, each of them relying on different features in order to select the optimal sperm. BINDING ASSAY TECHNIQUES Physiological intracytoplasmic injection (PICSI) This technique is based on the fact that hyaluronic acid (HA) plays an important role in selecting functionally competent sperm during in vivo fertilization (5). Not only HA is the main component of the cumulus matrix that surrounds the human oocyte, but also a natural selector of developmentally mature sperm. Furthermore, experimental data have shown that it can be used with a similar effect in vitro. HA-binding spermatozoa in vitro exhibit complete plasma membrane remodelling, decreased levels of cytoplasmic inclusions and residual histones, nuclear maturation and reduced risk of chromosome imbalance (i.e. chromosomal aneuploidy) and chromatin diseases (4). Currently, there are two options available to perform this technique. First, plastic culture dishes with HA hydrogel microdots attached to the bottom. After using this method the desired spermatozoa remain bound to the microdots by the head, while flagella beat in place (4). Secondly, the use of an alternate HA-containing viscous medium, method known as “Sperm Slow” because the spermatozoa appear “slowed down” (4). Since PICSI is considered to better assess the physiological potential in order to select sperm for ICSI, some authors claim it should replace the use of conventional ICSI and potentially toxic polyvinylpyrrolidone (PVP) (used to slow down sperm movement). Nevertheless, significant improvement of IVF outcomes using HA-based sperm selection is still to be confirmed in larger studies (6, 7), reason why the use of PICSI is currently limited to punctual cases in which significant improvement of clinical outcomes has been proved (8). Such cases include total fertilization failure by ICSI, high levels of DNA fragmentation, impaired embryo development, failure to implant in the uterus and recurrent miscarriages (9, 10). Fertilization failure after ICSI can sometimes be explained by problems in sperm nucleus decondensation and chromatin abnormalities related to DNA fragmentation (11). Sperm with such DNA damage and/or chromosome unbalance are avoided by PICSI/HA binding negative selection (3). Although it is a controversial issue, several authors have found a significant improvement of embryo quality and implantation ability when performing PICSI vs conventional ICSI, as well as a reduced number of miscarriages (3, 11, 12, 13, 14). The decreased incidence of miscarriages seems to be linked to the incapacity of sperm with genetic defects to form a normal pro-nucleus after conventional ICSI, thus generating aneuploid embryos with high levels of fragmentation during cleavage. Such aberrant embryos normally end up in pregnancy loss (14). Since PICSI allows for selection of sperm with properly compacted chromatin, embryos derived from these spermatozoa are less likely to present defects in quality (15). This translates into a higher implantation potential and a lower incidence of miscarriage, which eventually improves the reproductive outcome (12, 13). Magnetic activation cell sorting (MACS) In 2008 Said and colleagues proposed a protocol combining a new method, denominated magnetic activation cell sorting (MACS), and density gradient column (DGC) in order to obtain higher-quality sperm samples (2). MACS consists in mixing up the semen sample with annexine V (A5) (known to have high affinity with phosphatidylserine (PS)) coupled to magnetic microspheres. The sample is then exposed to a magnetic field in an affinity column (16). A5-bound sperm have been linked to PS externalisation, a well-known sign of apoptosis due to the loss of membrane integrity. MACS should be performed prior to DGC because PS externalisation naturally occurs during sperm capacitation by DGC, and so MACS may inadvertently discard good-quality sperm (17). During the last decade, several studies have corroborated the efficient use of this protocol in order to obtain sperm samples with a low DNA fragmentation index and higher fertilization potential (2, 16). Because MACS reduces the percentage of apoptotic sperm selected from the sample, this technique is mostly indicated in cases of high rates of DNA damage or predisposition to suffer from it (18). Such cases include: patients with altered sperm parameters, cryopreserved spermatozoa from cancer patients (19), previous fertilization failure, recurrent implantation failure and recurrent miscarriage (20, 21). Several clinics have implemented MACS in their daily routine, claiming selection of sperm through MACS to improve their ICSI outcome (22, 23, 24). However, such statement remains controversial, since other authors have reported no actual beneficial effect after using this technique (16, 25). Zona binding This method mimics natural sperm selection using the zona pellucida (ZP) from an immature sibling oocyte (26). The sperm sample is processed by DGC and then incubated along with the ZP for two hours. ZP-bound sperm are then eligible for ICSI. Although various studies have linked the ZP binding assay prior to ICSI to improved embryo quality and implantation rate, it is still not feasible to employ this method on a routine basis due to the extra amount of work and time required. Therefore, it is currently recommended to remain restricted to patients with poor outcomes in previous ICSI cycles, or to those who exhibit sperm with damaged DNA or abnormal morphologies (2). MICROSCOPY BASED TECHNIQUES Intracytoplasmic morphologically selected injection (IMSI) Studies have shown that sperm classified as morphologically “normal” at conventional optical resolution and magnification (x200-x400) may carry ultrastructural defects. Many of these abnormalities have been linked to hidden chromosomal defects, high levels of DNA fragmentation, abnormal centriolar function, etc. (1, 2). These aberrations may impede fertilization, lead to post-fertilization arrest or even disrupt embryo development (2). IMSI is an approach developed from a method of sperm evaluation, first described in 2002 by Bartoov and colleagues and denominated “motile sperm organelle morphology examination” (MSOME) (27). In order to apply this evaluation technique, observations need to be performed at x6000-6600 magnification (1). Performance of sperm selection along with MSOME requires the use of specific equipment. This should be an inverted light microscope with high-power optics intended for differential interference contrast and enhanced by digital imaging, which allows the embryologist to assess sperm morphology in real time. Sperm selection is carried out in a dish different from the one used for ICSI; it is a glass-bottomed dish that allows for the best optimal quality. Semen samples for IMSI are subjected to discontinuous DGC beforehand, but the exact procedure may vary depending on the quality of the sample (2). Sperm selection based on IMSI—MSOME findings helps to discard spermatozoa with mitochondrial dysfunction or DNA damage (4). In this regard, IMSI-MSOME has been proved useful for oligo-asthenoteratozoospermic patients (28) and for couples with recurrent implantation failures, reducing miscarriage rates by 50% (29). Nevertheless, simpler techniques are available for selecting sperm with low DNA fragmentation such as PICSI and MACS, so IMSI-MSOME is not yet extendedly used (4). In fact, this technique did not have the expected impact due to its downsides: high cost and long time for selecting sperm for ICSI. Depending on semen quality, selection may take around 60-120 minutes, which could negatively affect cells given that sperm nuclei may vacuolize after 2-hour exposure to warm media (30). Polarized light microscope The different anisotropic properties of spermatozoa through its protoplasmic texture provoke polarized light to be refracted at different speeds. These differences between refractions are known as retardance or birefringence (31). Viable human spermatozoa are naturally birefringent, while this characteristic is absent in pathological, dead or necrotic ones due to changes in the molecular structures of the cell and/or organelles (nucleoprotein filaments, axoneme, mitochondria, etc.) (32, 33). Examined under polarized microscope (PM), a normal spermatozoon will exhibit a non-luminous acrosome and luminous and normal-sized compact nucleus and flagellum (Giulia Collodel 2010). Multiple studies support the correlation between sperm head birefringence and DNA fragmentation, fertilization rates and higher embryo quality (2). The increase of a sperm head retardance is positively correlated with DNA damage, the optimal value for sperm head retardance oscillates between 0.56 nm and 0.91 nm. (32). Thanks to improvements in polarized light microscopy it is now possible to evaluate viability, motility, morphology and concentration of a sperm sample, all in a single step and with no need of exposure to potentially harmful dyes or environmental conditions (33). MEMBRANE POTENTIAL BASED TECHNIQUES Zeta potential method Zeta potential refers to the negative electric potential observed between the outer medium and the sperm membrane surface. The sperm selection technique based on this potential was developed following the observation that sperm with damaged DNA exhibited a lower Zeta potential (34). This method consists in the use of positively charged test tubes to which morphologically normal spermatozoa containing intact DNA can bind (2). This promising, easy and time-efficient new method presents the major drawback of a low recovery rate (8.8%), which is especially troubling in cases of oligozoospermic patients, who represent a high percentage of ICSI candidates (34). Micro-electrophoresis During sperm maturation in the epididymis, capacitation and acrosome reaction, the sperm membrane undergoes specific modifications (35, 36), among which the addition of sialic acid residues is noteworthy. High concentrations of this residue reflect normal spermatogenesis and sperm maturation (37), as well as higher negative charge compared to immature or abnormal sperm (38). Taking advantage of such trait, negatively-charged spermatozoa are selected as they migrate towards the anode during electrophoretic sperm separation (39). Similarly to the Zeta-potential method, micro-electrophoresis enables the isolation of sperm with no DNA damage, since they present higher negative net charge (40). Although micro-electrophoresis remains an experimental method, the percentage of negatively charged sperm is directly associated with fertilization rate and blastocyst development and inversely associated with embryo arrest (41). ABSOLUTE IMMOTILE SPERM SELECTION TECHNIQUES: There are some cases of severe male factor infertility, such as total necrozoospermia, in which absolute immotile sperm are prevalent. In such cases, testicular sperm extraction (TESE) seems to be the only actual solution; however, obtaining motile and/or viable sperm for ICSI may result an impossible goal (42). Difficulties to select viable sperm can also occur in cases of severe cryptozoospermia and asthenozoospermia. Therefore, several techniques have been developed in order to differentiate immotile and non-viable sperm (2, 43, 44, 45). Hypo-osmotic swelling test (HOST) Studies have shown that in cases of severe male infertility normal sperm morphology will still likely have a high DNA fragmentation index. HOST is a method to estimate chromatin integrity (2), based on the degree of swelling of the cytoplasm and curling of the tail in live sperm when exposed to hypo-osmotic conditions. Different swelling patterns correspond to different degrees of chromatin integrity; by identifying these patterns, live spermatozoa with normal membrane function and low DNA fragmentation can be selected (2, 43). Laser assisted immotile sperm selection (LAISS) LAISS helps to differentiate between viable and dead spermatozoa by evaluating curling of the flagellum, as detected in live sperm when hit by the laser. The use of LAISS has reported results comparable to HOST; however, the former is much quicker than the latter, and its effect can be immediately observed without the need of continued evaluation, contrary to what is required for HOST (43). Mechanical touch technique or the sperm tail flexibility test (STFT) Although HOST is the most used test to differentiate live immotile spermatozoa from dead ones, evidence suggests that it may not be totally reliable (46, 47). Furthermore, solutions used for hypo-osmotic shock can be harmful for spermatozoa. In 2003, Soares and coauthors published their results using a simple and low-cost technique for selecting viable immotile sperm. They observed acceptable fertilization rates after injecting spermatozoa with flexible tail from total immotile sperm samples. Based on these results, the authors proposed STFT as a means to discriminate live from dead sperm; STFT consists in touching the flagellum with the ICSI pipette to check flexibility. Spermatozoa with flexible (non-stiff) flagellum may be considered alive and therefore eligible for ICSI (44). Methylxanthines: Pentoxifylline (PTX) Use of Pentoxifylline (PTX) enhances sperm motility by inhibiting the breakdown of cAMP which is essential for sperm motility (48). PTX is a 3’5’-nucleotide phosphodiesterase inhibitor that might result highly toxic for the oocyte or the embryo (2). Nevertheless, due to the reported increased fertilisation rates for immotile sperm after exposure to PTX (48), this method is still recommended for thawed testicular sperm samples, though only in cases of 100% immotile sperm, and always for short periods of time (2, 48). ATP/MgSO4 In 2014 Neri and colleagues proposed the use of ATP/MgSO4 solution for immotile spermatozoa that did not respond to motility enhancers (49). It is especially recommended for surgically retrieved or thawed sperm samples (49). The exposure to ATP/MgSO4 stimulate sperm kinetic machinery provoking viable, but immotile spermatozoa flagella to twitch (2). The same team reported that 64.6% of spermatozoa from an only immotile sperm sample exhibited flagellar movement after exposure to ATP/MgSO4 (49). CONCLUSIONS ICSI is a highly efficient technique, which reaches up to 85-90% fertilisation rates. Two main causes can explain fertilisation failure by this technique: the lack of appropriate sperm for injection or failed oocyte activation. Having covered the latter in a previous publication, the present post aims to review the current available techniques for the improved sperm selection for ICSI. Among these, IMSI, PICSI and MACS are widely studied, and their efficiency extensively accepted. Several novel approaches have been also developed. HOST, polarised microscopy and micro-electrophoresis are suggested to improve fertilisation rates, although multicenter randomised control trials will be needed before their general implementation to IVF routine. On the other hand, ZP binding assay and Zeta potential method are still experimental techniques that are yet to demonstrate their clinical advantages and feasibility. Finally, the best method for selecting live spermatozoa from totally immotile sperm samples remains a controversial issue. Whereas HOST seems to be the most extended one, simpler and easier techniques have appeared in the last decade that may also prove highly useful for an effective good-quality sperm selection. REFERENCES
Authors: Belén Gómez Giménez and Edel Rocher "The assessment of embryo development in vitro suggests that collapsing of the blastocoel might be indeed related to a certain degree of success" 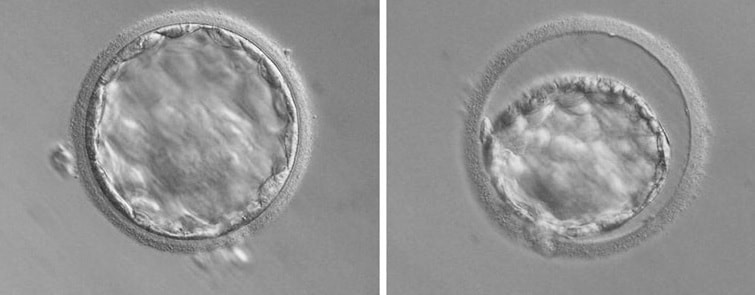 Figure 1. Artificial shrinkage of blastocoelic cavity in human expanded blastocysts by hyperosmotic sucrose solutions, before and after the treatment (1). One of the main factors related to embryo selection for transfer is the extension of embryo culture up to blastocyst stage. This approach has been demonstrated to improve clinical outcomes after in vitro fertilization IVF (2). Consequently, in the last couple decades a specific blastocyst grading system has been applied to assess embryo morphology during blastocyst stage (appearance of the inner cell mass (ICM) and the trophectoderm (TE)) [read our post on blastocyst assessment]. But there are other aspects about blastocyst development to focus on, such as the study of collapse and re-expansion in vitro and how it could affect implantation and pregnancy rates. BLASTOCYST COLLAPSE UNDER NORMAL CIRCUMSTANCES The term "collapse" refers to the contractions observed in the blastocyst that respond to a series of physical phenomena related to its proper development. Once morula stage has been reached, the forming blastomeres begin to pump ions through the Na+/K+ machinery (3). This entails an implicit osmotic response that results in the accumulation of water in the cavity thus formed, the blastocoel. The progressive accumulation of water causes the blastocyst to grow in size from early stages, consequently increasing the hydrostatic pressure between the TE and the zona pellucida (ZP) until the blastocyst hatches (4, 5). The efflux of the blastocoel fluid through loose cell bindings in the already formed TE causes the aforementioned contractions or "collapse". This phenomenon was originally called "blastocyst breathing", due to the sequence of collapse and re-expansion events observed in the rupture of the ZP during blastocyst hatching (6). However, the mechanisms of blastocoel collapse and TE recovery after the rupture in vitro still remain unclear (7). INFLUENCE OF THE NATURAL COLLAPSE OF BLASTOCYSTS ON REPRODUCTIVE OUTCOMES When considering the effect of blastocyst collapse on future outcomes, certain aspects must be taken into account: 1. Number of collapsing episodes Nowadays, there are still few studies on the frequency of blastocyst collapse. Marcos and coauthors (2015) focused on this aspect for the first time and reported almost a 20% of single collapse episodes in a total of 715 blastocysts, whereas only about 1.5% exhibited multiple collapse events (8). Independent authors have further studied this topic; even though they all have shown significant variations in the percentages of both single and multiple collapse events during blastocyst stage (8, 9), results do suggest a correlation between the number of collapsing episodes and the future outcome for the blastocyst (7, 8, 9). 2. Effect of collapse on blastocyst hatching Research on certain mammalian species had shown that blastocysts with smaller contractions (collapse/re-expansion events) were more likely to reach the hatching stage than those with large strong contractions (10, 11, 12). Results in patients showed that differences in hatching rates were not statistically significant between blastocysts with and without collapse (28.7% vs 31%, respectively) (7). However, hatched embryos with previous collapse episodes exhibited lower implantation rates than those with no collapse (35.1% vs 48.5%, respectively) (8). Taken together, these results indicate the process of collapse and re-expansion may not have a direct influence on blastocyst development up to the hatching stage, but they seem to negatively affect the success rate of such blastocysts after implantation in the uterus. 3. Relationship between standard morphological evaluations and blastocyst collapse Morphological assessment of embryos has also been used in order to figure out the main cause for blastocyst collapse. This evaluation has been performed considering the number of blastomeres, level of fragmentation and quality in days 2, 3 and 5 (blastocyst stage). Nevertheless, the comparison between all these standard morphological features did not yield significant differences that could relate to the event of blastocyst collapse (6, 8). Whereas further evaluation is a usual practice for non-hatched blastocysts, these studies showed no results regarding day 6. Although comparisons including this factor may potentially reveal connections between collapsing of blastocysts and their early developmental history, no data are currently available on this issue. 4. Prediction value of morphokinetic variables The evaluation of morphokinetic variables has proven to be useful in selecting embryos for transfer, given their potential to predict the successful development of the embryo. These variables have been associated with blastocyst formation, implantation potential and aneuploidy status (13, 14, 15), measured in different critical steps of embryo development (16). In spite of the increasing amount of studies focused on the relation between morphokinetic values and embryo implantation rates, only a few studies can be found that link these variables with the occurrence of blastocyst collapse (7, 9). Marco et al (2015) showed a significantly slower development of those embryos that had not collapsed, compared to those that did, considering development times from 2-cell stage up to blastulation (t2 - tB, respectively) (7). These results are found difficult to combine with those by Bodri and colleagues (2016); although the authors did relate blastocyst collapse to a progressively decreased live birth rate, they were not able to state this feature as a valid predictor of embryo survival on its own (9). ARTIFICIAL SHRINKAGE As it has been previously discussed, blastocyst collapse is a natural phenomenon linked to normal blastocyst development. Even though previous data have shown no direct correlation between this feature in vivo and implantation rates, recent and current studies in vitro suggest that the occurrence of blastocyst collapse has indeed a significant effect on the clinical outcome of the embryo, as explained below. Currently, vitrification is a widely spread practice in laboratories, aimed to preserve cells/tissues/organs at ultralow temperatures (-196°C). It is well known that the traditional slow freezing approach for cryopreservation frequently leads to ice crystal formation that may damage the cell/tissue, thus decreasing its quality. This associated issue has been avoided by the addition of high concentrations of cryoprotectants and a significant rise in the speed of temperature drop. Such a combination increases the viscosity of the solution and turns it into a glass-like structure (17). Supporting evidence of the success of this technique are numerous studies that demonstrate that the quality of vitrified embryos is comparable to the quality of fresh ones [find more about the success of frozen blastocysts on our previous post here]. A number of researchers have concluded that blastocoel size at the moment of vitrification has an effect on embryo survival and implantation rates. The authors postulated that a large fluid-filled cavity in expanded blastocysts may inhibit sufficient permeation of the cryoprotectant into the blastocoel, thus allowing ice formation and decreasing the chances of survival (18, 19). There exist several ways to reduce the size of the blastocoel in expanded blastocysts (D5-D6) through artificial shrinkage (AS). Some of them are summarised below:
In 2002, Vanderzwalmen and collaborators achieved AS by introducing a needle into the blastocoel until blastocyst contraction was observed (18). However, and even though the authors reported an increase in survival and implantation rates compared to intact blastocysts, the increase in pregnancy rates was not statistically significant. Similar results were achieved by Son et al (2003), who induced AS prior vitrification, obtaining about 90% of blastocyst survival. Having implanted almost a third of the total, 48% of those turned into a clinical pregnancy (21). However, these studies were regarded as invasive, due to the injury the method caused in the ZP and the TE. In 2004, Hiraoka et al reported AS of the blastocoel by mechanical pipetting using a fine hand-drawn glass pipette. Even though this method also harms the TE, consequences are less severe due to the needles used. These authors also observed increased survival and pregnancy rates after AS (22). 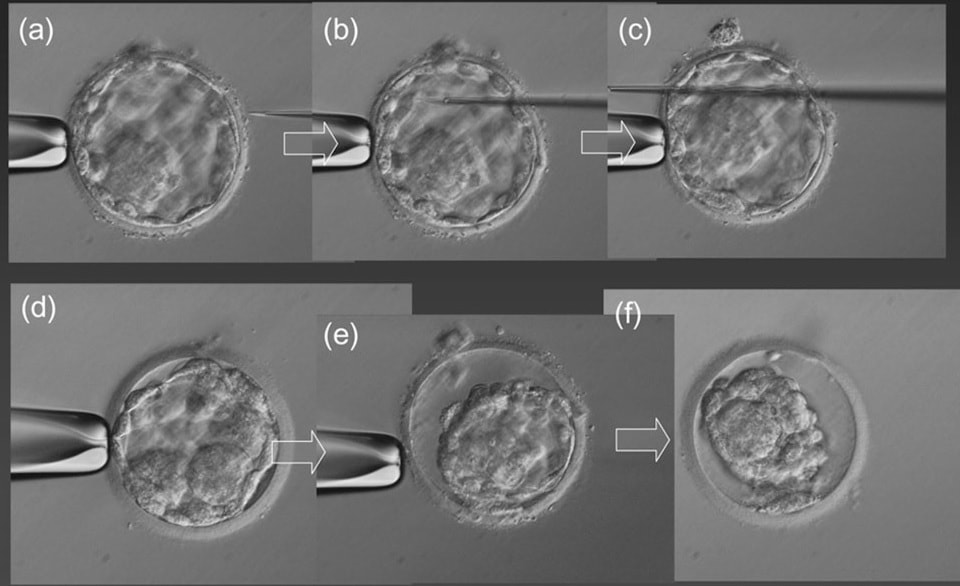 Figure 2. Artificial shrinkage (AS) of expanded blastocyst with the micro-needle: (a) holding the expanded blastocyst with the holding micropipette. (b) Insertion of the micro-needle inside the blastocoel at a point away from the ICM. (c) Puncture through the blastocoel and gradual removal of the micro-needle. (d) Beginning of shrinkage 10 s after puncture. (e) Partial shrinkage 30 s after puncture. (f) Complete shrinkage 1 min after puncture. Magnification is ×400 (23). Laser pulse has been also previously used for AS. Application of this method has reported survival rates to reach 97% and pregnancy rates up to 60% (23). In a randomized study, Van Landuyt et al (2015) found no significant increase in implantation rates after applying the pulse prior vitrification, but they did report higher post-warming blastocyst survival rate and quality in collapsed blastocysts (19). After AS, blastocysts were more likely to reach the hatching stage than those not subjected to the procedure. Similar results were obtained by Darwish et al (2016), who observed improved blastocyst survival, clinical pregnancy and implantation rates after removal of the blastocoel fluid (24). In addition, artificially collapsed blastocysts before vitrification have been recently found to re-expand more rapidly after warming (25). However, differences in live birth rates were not significant and the sample size used in the study may have been too small, so these results should be taken carefully. Other methods have been employed for AS, too. In fact, compared studies have been performed to assess differences on the protocols that may result in improved clinical outcomes. Laser pulses applied on cell-to-cell junctions on the TE and exposure of blastocysts to hyperosmotic sucrose solutions both provided evidence of increased probability of fast-developing embryos and higher implantation rates (1). Even though the authors found no significant differences between methods, the latter was suggested based on the cost/benefit ratio. These and similar results suggest that AS of human expanded and hatching blastocysts is actually a useful approach to improve clinical outcomes, regardless of the methodology employed. However, and even though it seems that collapse of the blastocoel might be indeed somehow linked to higher implantation and/or pregnancy rates, conclusions should be still drawn carefully. CONCLUSIONS Blastocyst collapse has been found to be an inherent trait of embryo early development. Its occurrence seems to be related to the physical normal progression of the embryo, and depending on the normal microenvironmental conditions, starting at the zygote stage and expanding up to the last moments before hatching and implantation. Whereas there is no compelling evidence of a link between blastocyst collapse and implantation success in natural pregnancies, the assessment of embryo development in vitro suggests that collapsing of the blastocoel might be indeed related to a certain degree of success. Comparisons between different AS methods have revealed different behaviour of blastocysts after implantation, and even an increasing number of collapsing episodes may be related to a decreased implantation potential. It is also worth to be noted the importance of vitrification in the routine practice in the laboratory; even though this process optimizes cryopreservation of cells and tissues, the integrity of the embryo may result compromised. Should this be true, blastocoel collapse may become more evident after warming of the embryos, or simply these blastocysts are more prone to collapsing, thus existing correlation rather than causation between both events. Because blastocyst collapse has been payed attention to in laboratories for just a few years now, thorough and more insightful studies are required to clarify a real connection between this feature and the outcomes of clinical pregnancies. REFERENCES
(1) Iwayama H, Hochi S, Yamashita M. In vitro and in vivo viability of human blastocysts collapsed by laser pulse or osmotic shock prior to vitrification. J Assist Reprod Genet. 2011;28(4):355-61. (2) Blake D, Farquhar CN, Johnson N, Proctor M. Cleavage stage versus blastocyst stage embryo transfer in assisted conception. Cochrane Database Syst Rev 2007; 4: CD002118. (3) Baltz JM, Smith SS, Biggers JD, Lechene C. Intracellular ion concentrations and their maintenance by Na+/K(+)-ATPase in preimplantation mouse embryos. Zygote 1997; 5: 1–9. (4) Biggers JD. Reflections on the culture of the preimplantation embryo. Int J Dev Biol 1998; 42: 879–884. (5) Watson AJ, Natale DR, Barcroft LC. Molecular regulation of blastocyst formation. Anim Reprod Sci 2004; 82–83: 583–592. (6) . Veeck LL, Zaninovic N. Human blastocysts in vitro. In: Veeck L, Zaninovic N (eds). An Atlas of Human Blastocysts. Parthenon Publishing 2003; 99–137. (7) Marcos J, Pérez-Albalá S, Mifsud A, Molla M, Landeras J, Meseguer M. Collapse of blastocysts is strongly related to lower implantation success: a time-lapse study. Human Reproduction 2015; 30 (11); 2501–2508. (8) Esbert M, Marconetto A, Soares SR, Quera M, Molina J, Florensa M, Ballesteros A, Meseguer M. Does the blastocyst collapse respond to a biological need? The analysis of 1,952 embryos by time-lapse can give an answer. Fertil Steril 2017; 108 (3, Supplement):e157. (9) Bodri D, Sugimoto T, Yao Serna J, Kawachiya S, Kato R, Matsumoto T. Blastocyst collapse is not an independent predictor of reduced live birth: a time-lapse study. Fertil Steril. 2016; 105(6): 1476-1483. (10) Erbach GT, Biggers JD, Manning PC, Nowak RA. Localization of parathyroid hormone-related protein in the preimplantation mouse embryo is associated with events of blastocyst hatching. J Assist Reprod Genet 2013; 30: 1009–1015. (11) Gonzales DS, Jones JM, Pinyopummintr T, Carnevale EM, Ginther OJ, Shapiro SS, Bavister BD. Trophectoderm projections: potential means for locomotion, attachment and implantation of bovine, equine, and human blastocysts. Hum Reprod 1996; 11: 2739–2745. (12) Seshagiri PB, Sen Roy S, Sireesha G, Rao RP. Cellular and molecular regulation of mammalian blastocyst hatching. J Reprod Immun 2009; 83: 79–84. (13) Cruz M, Garrido N, Herrero J, Perez-Cano I, Munoz M, Meseguer M. Timing of cell division in human cleavage-stage embryos is linked with blastocyst formation and quality. Reprod Biomed Online 2012; 25: 371–381. (14) Dal Canto M, Coticchio G, Mignini Renzini M, de Ponti E, Novara PV, Brambillasca F et al. Cleavage kinetics analysis of human embryos predicts development to blastocyst and implantation. Reprod Biomed Online 2012; 25: 474–480. (15) Campbell A, Fishel S, Bowman N, Duffy S, Sedler M, Hickman CF. Modelling a risk classification of aneuploidy in human embryos using non-invasive morphokinetics. Reprod Biomed Online 2013; 26: 477–485. (16) Meseguer M, Herrero J, Tejera A, Hilligsoe KM, Ramsing NB, Remohi J. The use of morphokinetics as a predictor of embryo implantation. Hum Reprod 2011; 26: 2658–2671. (17) Armitage WJ. Cryopreservation of animal cells. Symp Soc Exp Biol. 1987;41:379-93. (18) Vanderzwalmen P, Bertin G, Debauche C, Standaert V, van Roosendaal E, Vandervorst M et al. Births after vitrification at morula and blastocyst stages: effect of artificial reduction of the blastocoelic cavity before vitrification. Hum Reprod. 2002;17(3):744-51. (19) Van Landuyt L, Polyzos NP, De Munck N, Blockeel C, Van de Velde H, Verheyen G. A prospective randomized controlled trial investigating the effect of artificial shrinkage (collapse) on the implantation potential of vitrified blastocysts. Hum Reprod. 2015;30(11):2509-18. (20) Bedient CE, Khanna P, Desai N. Laser Pulse Application in IVF. In: InTech. Lasers - Applications in Science and Industry. Las Vegas. 2011. p. 204. (21) Son WY, Yoon SH, Yoon HJ, Lee SM, Lim JH. Pregnancy outcome following transfer of human blastocysts vitrified on electron microscopy grids after induced collapse of the blastocoele. Hum Reprod. 2003;18(1):137-9. (22) Hiraoka K, Kinutani M, Kinutani K. Blastocoele collapse by micropipetting prior to vitrification gives excellent survival and pregnancy outcomes for human day 5 and 6 expanded blastocysts. Hum Reprod. 2004;19(12):2884-8. (23) Mukaida T, Oka C, Goto T, Takahashi K. Artificial shrinkage of blastocoeles using either a micro-needle or a laser pulse prior to the cooling steps of vitrification improves survival rate and pregnancy outcome of vitrified human blastocysts. Hum Reprod. 2006;21(12):3246-52. (24) Darwish E, Magdi Y. Artificial shrinkage of blastocoel using a laser pulse prior to vitrification improves clinical outcome. J Assist Reprod Genet. 2016;33(4):467-71. (25) Kovačič B, Taborin M, Vlaisavljević V. Artificial blastocoel collapse of human blastocysts before vitrification and its effect on re-expansion after warming - a prospective observational study using time-lapse microscopy. Reprod Biomed Online. 2018;36(2):121-9. Authors: María Caballero & Lidón Carretero Vilarroig "The main cause for unsuccessful ICSI seems to be failure in oocyte activation mechanisms" 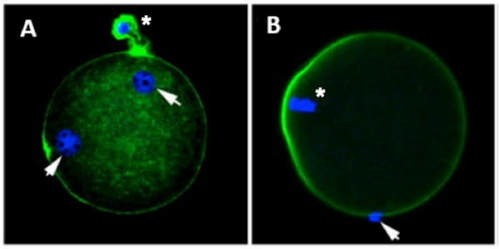 Fig 1. Zona-free mouse oocytes prepared for IVF. Staining with Draq 5 for DNA (blue) and phalloidin-Alexa Fluor 488 for filamentous actin (green) has been performed. (A) An oocyte with two pronuclei (arrows) and a polar body (asterisk) is shown, meaning activation has already begun after fertilization. (B) A spermatozoon (arrow) is observed bound to the oocyte not having penetrated the cortical actin. The equatorial plate in the meiotic spindle (asterisk) suggests the oocyte remains arrested at MII (polar body has presumably been lost during oocyte denudation), which indicates that oocyte activation has not yet been produced, given fertilization has actually not been achieved (modified from [1]). INTRODUCTION Fertilization involves the fusion of male and female gametes. However, for this to occur both cells must undergo certain morphological and physiological changes in order to be able to fuse. The sperm cell must undergo a process known as capacitation, which implies a series of changes regarding motility and plasma membrane composition (among others), essential to acquire the ability to fertilize. The oocyte in turn must not only complete several molecular changes involved in maturation, but also a combination of processes initiated by the sperm entry that culminate in the so-called oocyte activation (OA), which prepares the newly formed zygote for embryogenesis (2). In the context of in vitro fertilization (IVF) cycles, oocyte activation failure (Fig. 1) has been observed to occur in about 1-3% (3), resulting in visibly identifiable non-fertilized oocytes. In order to overcome this problem, assisted oocyte activation (AOA) has recently gained interest for those couples with a history of fertilization failure through intracytoplasmic sperm injection (ICSI). To better understand oocyte activation failure and AOA, here we review gamete maturation, focusing on oocyte activation mechanisms and where/when main problems may arise that cause failure in this process. GAMETE PREPARATION FOR FERTILIZATION Sperm Fresh ejaculated sperm is still not capable to fertilize. In order to be able to interact with the oocyte, the spermatozoon needs a final maturation step known as capacitation. This process takes place into the female reproductive tract, culminating in the acrosome reaction (4). During capacitation, the sperm cell undergoes biochemical changes related to the acquisition of fertilization ability, mainly involving the molecular reorganization and hyperpolarization of the plasma membrane, as well as hyperactivation (5). Changes in membrane organization are necessary in order to increase its fluidity, which is achieved by removing cholesterol. The mechanisms related to the efflux of cholesterol are not well understood yet, but albumin and high-density lipoproteins located in the female genital tract have been recently pointed out as the main responsible actors for this process (6). 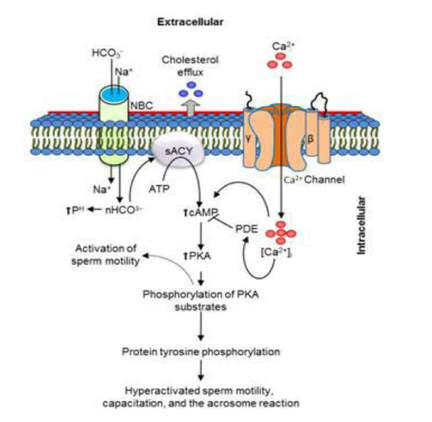 Fig 2. Molecular changes in spermatozoa responsible for motility activation, capacitation and acrosome reaction (7). Sperm membrane reorganization leads to the exposure of some important channels and receptors, which increases the permeabilization of sperm to certain ions. Some of the most important channels are CaSper and NBC, Ca2+ and Na+/HCO3- channels, respectively. Calcium influx plays an important role in the process of capacitation, contributing to acrosomal reaction and aiming to the exocytosis of the acrosomal vesicle (8). Another milestone of capacitation is hyperactivation, referred to as the acquisition of increased and differential sperm motility. This is activated by phosphorylation of protein kinase-A (PKA) in a (HCO3−)- and (Ca2+)-dependent manner, being these ions transported through the cell membrane via CaSper channel and NBC cotransporter, respectively (9). Additionally, tyrosine phosphorylation levels of a multitude of proteins rises significantly in the sperm while in the female tract. Such an increment causes an increase in the activity of the adenylate cyclase, which in turn causes intracellular cyclic adenosine monophosphate (cAMP) levels to rise, ultimately resulting in a differential oscillating movement of the flagellum (hyperactivation). This particular pattern of flagellar beating provides the sperm cell with the proper motility to move forward towards the ovum (Fig. 2). Finally, when the sperm cell reaches the oocyte, acrosome reaction (AR) is triggered, upon interaction with the zona pellucida (ZP). AR refers to the regulated exocytosis of the acrosome content (including several crucial enzymes) and exposure of surface antigens necessary for oocyte-sperm recognition. The acrosome and plasma membranes fuse, tipping out the content and enabling lateral contact between the sperm head plasma membrane and the oolemma. This is followed by fusion of the plasma membranes of both gametes and delivery of the sperm nuclear material into the oocyte. Oocyte Oocytes begin meiosis during foetal development and arrest at a special diplotene stage of prophase I called dictyotene or dictyate, so women are born with a limited number of primary oocytes. There are two mechanisms used by the oocyte to achieve meiotic arrest. The first one can be explained due to the low activity of M-phase promoting factor (MPF), promoted by the oocyte itself via endogenous production of cAMP (10). The oolemma exposes G-protein receptor (GPR3) leading to G-protein-coupled GPR3 and GPR12 activation (11), which in turn stimulates cAMP, a cyclase responsible for AMP synthesis. The second mechanism uses the cumulus-oocyte complex (COC) as a barrier that prevents meiosis reactivation. COC cells produce guanosine 3′, 5′-cyclic monophosphate (cGMP), which enters into the oocyte through gap junctions. Once inside the oocyte, cGMP inhibits the phosphodiesterase responsible for cAMP hydrolysis (PDE3A or cGMP-inhibited phosphodiesterase), which increases cAMP activity, thus inhibiting MPF (10) (Fig. 3). Primary oocytes remain at meiotic arrest until puberty, when the pituitary gland releases luteinizing hormone (LH) during the menstrual cycle and before ovulation. LH acts as a molecular key and activates MPF. LH receptors are located on the surface of granulosa cells, so they transfer the meiotic arrest release signal into the oocyte. Nevertheless, after the resumption of first meiotic division, the maintained activity of MPF promotes the oocyte arrest at metaphase II (12,13). 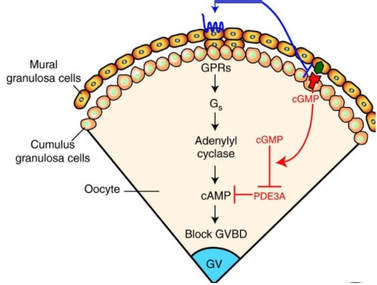 Fig 3. Signalling pathway of the regulation of oocyte maturation. At puberty stage, LH reverses the inhibition of meiotic maturation by decreasing the synthesis of cGMP (adapted from 11). OOCYTE ACTIVATION As a result of the sperm penetration into the oocyte, the female gamete undergoes a process called oocyte activation (OA). This refers to the resumption of the transcriptional activity of the oocyte (inactive until fertilization), essential to allow the subsequent embryo development to progress (14). OA is characterized by the triggering of several events, such as the resumption of meiosis, pronuclei formation, second polar body extrusion, cortical granule exocytosis and genetic material replication, among others (15,16). All these events are the result of an increase in intracellular calcium (Ca2+) concentration, which takes place upon gamete fusion. In mammals, this Ca2+ release is known as Ca2+ oscillations, due to the fact that this molecule is gradually released over time. Every species shows a specific pattern of frequency and duration for these oscillations (17). The “sperm factor” The idea of the sperm containing some kind of OA trigger has been on the table for decades. Evidences showing that sperm extracts are able to fertilize eggs from different species (even phyla) suggested the existence of a common factor that would act as a universal activator of the oocyte (18, 19). Independent observations from different models revealed that the so-called “sperm factor” could effectively be a soluble calcium releasing agent, role that has been attributed to several molecules to date, including phospholipases, second messengers and others. Even involvement of adenosin diphosphate ribose (ADPr) has been discussed in this context, since it has been shown to trigger calcium release through a nitric oxide-based pathway (20, 21). Other molecules such as citrate synthase (22) or post-acrosomal WWP-domain binding protein (PAWP) (23) have been considered potential candidates for years, after their involvement in triggering OA had been demonstrated. However, several pieces of evidence have shown that the key sperm factor responsible for intracellular Ca2+ release and the subsequent oocyte activation may be a testis-specific PLC isoform named PLCζ (24). Although PLCζ is mainly located in the spermatozoon head, it remains unclear if the triggering isoform localizes in the post-acrosomal, acrosomal or equatorial region (17). Once PLCζ is delivered into the ooplasm, it induces hydrolysis of phosphatidylinositol 4,5-biphosphate (PIP2), a membrane-bound substrate located in an intracellular vesicular membrane. The hydrolysis generates two second messengers: inositol triphosphate (IP3) and diacylglycerol (DAG). Thereupon, release of Ca2+ is induced by binding of IP3 to its receptor (IP3R), localized in the endoplasmic reticulum (ER) membrane. This molecular activity causes the characteristic Ca2+ pattern responsible for triggering subsequent events explained below (Fig. 4) (18, 25). Although IP3 causes the first Ca2+ oscillations, DAG has been reported to interact with protein kinase C (PKC), thus increasing its activity. PKC also seems to be involved in events such as the production of Ca2+ oscillations, meiotic resumption or triggering of cortical granule exocytosis (26). 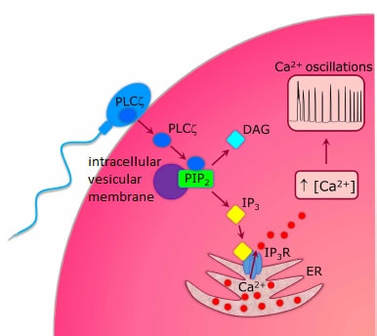 Fig 4. Schematic representation of egg activation triggered by sperm-specific PLCζ (modified from 17). The role of oocyte in activation Increased levels of intracellular Ca2+ have two consequences (Fig. 5). First, it enhances Ca2+ production by stimulating PLCζ and generating higher levels of IP3 (26). Secondly, it causes hyperpolarization of the plasma membrane, which results in the opening of specific channels present in the oolemma such as the store-operated Ca2+ entry (SOCE), the TRP family proteins and the plasma membrane (Ca2+)-ATPase (PMCA). Opening of these channels activates the influx of Ca2+, which is essential to keep Ca2+ oscillations and reload the intracellular stores (22, 28). 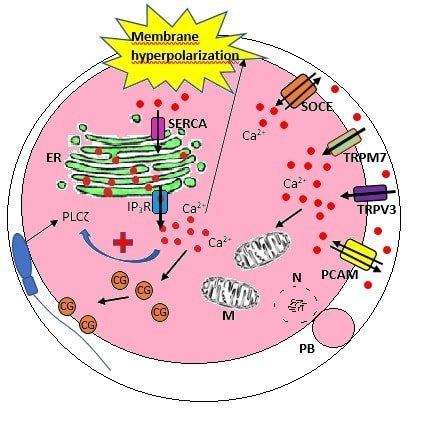 Fig. 5. Representation of the two consequences of Ca2+ increase in the fertilized oocyte. First, high Ca2+ concentration stimulates PLCζ activity and causes the hyperpolarization of the oolemma, which results in the opening of channels like SOCE, TRPM7, TRPV3 and PCAM. Secondly, whereas part of the released Ca2+ is retained by mitochondria, the rest enters into the ER through SERCA or leaves the oocyte through PCAM and SOCE channels. ER: endoplasmic reticulum. N: nucleus. M: mitochondria. CG: cortical granules. Ca2+: calcium. PB: polar body. Regarding intracellular Ca2+ release, it is necessary to keep a balance to prevent prolonged exposures to the ion that may be detrimental to the egg. Although part of this Ca2+ will be pumped back to the ER by sarcoplasmic/endoplasmic reticulum calcium-ATPase (SERCA) or the protein SOCE system, mitochondria are the actual major regulators of intracellular Ca2+ homeostasis, this means mitochondria retain Ca2+ during fertilization, so that they will act as a passive buffer regulating Ca2+ release. Ca2+ in turn activates oxidative phosphorylation (ATP synthesis). Moreover, it has been demonstrated that ATP generation may regulate intracellular Ca2+ release by making the IP3 receptors more sensitive to (Ca2+)-mediated activation (29). The remaining Ca2+ would then be expelled from the cell through PMCA and the Na+/Ca2+ exchanger (Fig.5) (30). "[...] the female gamete undergoes a process called oocyte activation (OA) [...] characterized by the triggering of several events, such as the resumption of meiosis, pronuclei formation [...] and genetic material replication, among others." Egg activation events As previously mentioned, the increase of intracellular Ca2+ concentration triggers certain key events that enable embryo development: Cortical granule exocytosis and changes in the ZP Once fertilization has occurred, the ZP needs to be modified in order to prevent binding and penetration of additional spermatozoa (“polyspermia”). This is achieved by releasing the content of the cortical granules (CGs), specialized secretory vesicles located in the periphery of the oocyte (31). First, CGs are translocated to the plasma membrane by two calmodulin-dependent proteins, CaMKII and MLCK. CaMKII is responsible for releasing the CGs attached to the cytoskeleton, whereas MLCK promotes CG translocation by phosphorylating the motor protein myosin II (14). Upon translocation, CG fuse with the oolemma in a calcium-dependent manner and deliver their enzymatic content to the perivitelline space (PS). The so secreted glycosidases, proteases and cross-linking enzymes modify the structural network of the ZP ("zona hardening"), thus hindering the entry of multiple sperm (14). Resumption of meiosis As aforementioned, MPF is directly involved in maintaining the MII arrest of human mature oocytes (32). MPF is a heterodimer composed of cyclin B and Cdc2 kinase, a regulatory and a catalytic subunit, respectively (33). Following fertilization, Ca2+ oscillations induced by PLCζ activate a phosphorylation cascade that eventually results in the destruction of cyclin B, the concomitant MPF deactivation and the subsequent resumption and completion of meiosis (14). Pronuclei formation and second polar body extrusion With the completion of meiosis, the secondary oocyte produces the second polar body and reorganizes its own chromosome complement in the form of a visible and spherical pronucleus. Formation of the paternal pronucleus in turn is dependent on maternal proteins; since the sperm genetic material is highly condensed due to its association with protamins (14), such condensation needs to be loosened in order to make DNA replication and gene expression possible. Maternal histones then replace protamins and the paternal pronucleus becomes appropriately remodelled. Then, maternal and paternal pronuclei fuse together in a process known as singamy or karyogamy (34). Maternal RNAs and zygote genome activation (ZGA) Upon egg activation, embryo development commences. However, during early stages, the embryo genome remains transcriptionally inactive. Therefore, the maternal mRNAs and proteins present in the oocyte take charge of early development (14). Maternal mRNAs are modified to regulate gene expression at the postranscriptional level, which includes edition, splicing, translation and degradation (35). Subsequently, during the so-called maternal-to-zygotic transition (MZT) nuclear genome becomes transcriptionally active. Two main molecular activities are involved in this period: (1) maternal clearance, which means the deletion of maternal mRNAs and proteins necessary for oocyte maturation and first stages of embryogenesis; and (2) gene expression with new instructions for embryo development. This process is activated by the maternal genome and it is known as zygotic genome activation (ZGA) (14). ZGA is not a sudden event, but it has been found to occur in a wave-like fashion. Similar studies in mice and humans have shown that ZGA commences as early as the 1-cell stage; concomitant with maternal mRNA degradation, a minor zygotic gene expression is observed, followed by the major ZGA and the mid-preimplantation gene activation (MGA) (compaction and cavitation processes are also accompanied by waves of gene expression activation) (36, 37). ICSI FOLLOWING OOCYTE ACTIVATION FAILURE Between 10-16% of couples worldwide are unable to have children without draw on assisted reproduction techniques (38). Both conventional IVF and ICSI have been proven to be highly successful in ART treatments worldwide, however, the use of ICSI has been reported to be of 66% in 61 countries between 2008 and 2010 and it continues increasing, even in cases with normal semen parameters (39). This success may be explained by to the fact that ICSI shows the highest success rates (around 97-99%) (38), and also because it represents a useful method to rescue fertility for men with suboptimal semen parameters, or even for couples whose fertilization rates after conventional IVF are close to zero (3). Despite the low failure rates, ICSI cycles do sometimes fail. There exist several possible causes that may explain failure of ICSI cycles, such as technical factors, failed sperm head decondensation, oocyte spindle defects or poor sperm chromatin condensation (40). Nevertheless, the main cause for unsuccessful ICSI seems to be failure in oocyte activation mechanisms, due to either sperm or oocyte defects (17, 39) (Table 1). Sperm factors associated with oocyte activation failure Several independent studies have found deficient PLCζ levels when both morphologically normal and abnormal sperm (such as in cases of globozoospermia) from different patients were analysed using mouse oocytes (41, 42). Results have shown reduced or absent levels of PLCζ in the sperm head to be associated with two mutations found in the active domain-coding regions of the gene. Although a deficiency in PLCζ has been associated with most cases of men infertility, other molecules might also be involved in oocyte activation failure. Evidences exist that have linked PAWP, a sperm-specific protein located in the sperm perinuclear theca, to the process of OA. Previous studies have shown this protein to induce pronuclei formation and meiosis resumption when injected into swine mature oocytes. The authors suggested one of PAWP domains to interact with PLCγ, a PLC isoform present in the oolemma, thus contributing to the generation of Ca2+ oscillations (23). This suggests that defective levels of the protein or defective interactions with oocyte factors may eventually lead to oocyte activation failure (23). Oocyte factors associated with oocyte activation failure However, neither PLCζ nor any other sperm factor deficiency is always the main responsible for ICSI failure. This points to the existence of oocyte factors that contribute to such failure, as well. Unfortunately, evaluation of human oocyte-related problems is usually quite difficult due to ethical issues and restrictions. As a consequence, the number of available oocytes destined for research is actually low. Therefore, an in-depth study of these potential oocyte-related factors is still necessary nowadays (39). Despite such problems, there are some actual hypotheses about oocyte-related factors that might be involved in activation failure. As it has been previously exposed, PIP2 plays an essential role, since it is the first element to interact with PLCζ. Some studies have suggested that either a small number of oocyte vesicles containing PIP2 or even a low amount of PIP2 in them could lead to oocyte activation failure (43). Once PIP2 is hydrolysed, IP3 binds to its receptor. Certain genetic mutations in IP3R may block this interaction, thus preventing Ca2+ oscillations to occur. Such mutations may also be responsible for abnormal protein folding or abnormal protein trafficking (39). Moreover, high levels of PLCζ are likely to lead to overproduction of DAG, which may contribute to oocyte activation failure by creating unusual patterns of Ca2+ oscillations (26). Even when Ca2+ oscillations occur normally, it is important to keep a balance within and outside the oocyte; any mistake in homeostasis regulatory mechanisms may compromise OA (39). As the main regulators of homeostasis, mitochondria function needs to be guaranteed; defective mitochondrial activity due to a low number of these organelles may alter ATP production and generation of Ca2+ oscillations, leading to defects in OA (44). On the other hand, homeostasis is also regulated by the correct function of specific membrane channels. For instance, SOCE is responsible for the introduction of Ca2+ into cellular stores (45). Both components of this system, STIMI and ORAII, may also result mutated, in which case their compromised function would negatively affect the normal occurrence of Ca2+ oscillations (39). There also exist other oocyte proteins that could lead to activation failure. For instance, the above-mentioned CaMKII is a calcium/calmodulin-dependent protein kinase II that induces cyclin B1 degradation, allowing resumption of meiosis (46). CaMKII is dependent on Ca2+ oscillations, so defects in calcium homeostasis might impair the proper function of the protein and, in consequence, the meiotic progression (39). Additionally, other PLC isoforms present in the oolemma and different from PLCζ like β, γ or δ also seem to have a role in Ca2+ oscillations; in particular isoform γ has been found to interact with PAWP (29, 47). 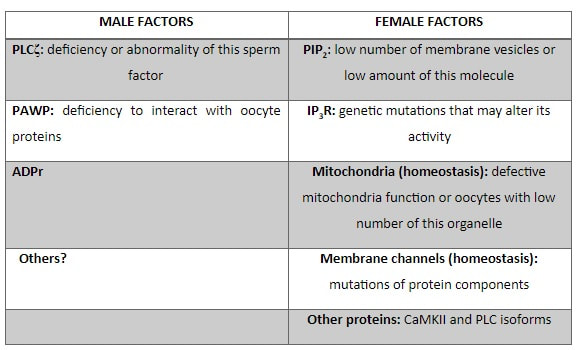 Table 1. Some of the most important factors potentially involved in oocyte activation failure. ASSISTED OOCYTE ACTIVATION Being oocyte activation failure the main cause for fertilization failure in ICSI cycles, some private clinics perform a special technique to test whether sperm is responsible for failure in oocyte activation. This test is known as MOAT test (Mouse Oocyte Activation Test) (48), and it consists in performing ICSI using sperm sampled from the patient and mouse oocytes. Sperm samples with proven fertility are used as positive controls, while the negative control is represented by injection of culture medium. Based on the results, patients are classified into one of three groups depending on the oocyte activation rate: (i) 0-20% (non-activation group); (ii) 21-84% (intermediate activation group); and (iii) 84-100% (sperm-related infertility refused). In addition, a calcium pattern analysis is also performed. Calcium plays a very important role in all forms of cell signaling. As already explained, calcium contributes to acrosomal reaction and enables the spermatozoon to penetrate into the oocyte (49). When the spermatozoon fertilizes the ovum, it triggers the generation of calcium waves leading to OA and embryonic development (50). A recent article published by Ferrer-Buitrago et al discusses whether calcium analysis can predict the response to AOA in patients with a history of failed fertilization after ICSI. Even though the sample size was limited, the results shown suggest that calcium analysis may actually foretell the response of ICSI-AOA, only in patients with abnormal capacity to generate Ca2+ oscillations (51). There have been several protocols used for AOA depending on the mechanisms used to trigger Ca2+ oscillations, which can be classified into electrical, mechanical or chemical (52). The first one actually involves electroporation of the oocyte, which generates micropores in the cell membrane allowing Ca2+ influx (53). The mechanical approach consists in oolemma breakage by vigorous aspiration of the ooplasm. The spermatozoon is then introduced into the oocyte, followed by energetic ooplasm aspiration and re-injection during ICSI. This way extracellular Ca2+ from the medium is introduced into the oocyte, resulting in its activation (54). Nevertheless, chemical-based protocols are the most extended techniques. Treatment with ionomycin, calcimycin, ethanol, strontium chloride (StCl2) or calcium ionophores can restore normal calcium oscillations (55). Among the later, the use of A23187 is extended about patients with globozoospermia, whose sperm lack the acrosome and may carry a mutant form of PLCζ. According to the protocol by Rybouchkin et al., oocytes are exposed to the ionophore in the IVF medium after ICSI, and cleavage stage can be assessed the next day (55, 56). Several cases of successful pregnancy have been reported following this ICSI-AOA approach. Few years ago, Hye Jin Yoon and coauthors published results from 185 ICSI AOA cycles using a calcium ionophore ending up in thirty-eight healthy babies with no congenital birth defects (57). The same year, Kim et al. (58) reported five successful pregnancies following ICSI-StCl2 treatment that resulted in the birth of eight healthy children. These results point to AOA as a new therapeutic tool for couples experiencing complete fertilization failure after ICSI. CONCLUSIONS Although ICSI cycles show a high percentage of success (97-99%), there is still certain probability of not achieving fertilization. Among the different causes responsible for this, the most common is the oocyte activation failure. For fertilization to succeed, it is important that both female and male gametes undergo a series of changes. In the case of spermatozoa, they need to be capacitated before oocyte penetration. On the other hand, upon fertilization, the mature oocyte needs to be activated in order to start embryo development. This process, known as oocyte activation, is characterized by internal Ca2+ oscillations produced by sperm factors like PLCζ, which is liberated into the ooplasm after fertilization. Ca2+ oscillations trigger the following events: (1) resumption of meiosis, (2) pronuclei formation, (3) second polar body extrusion, (4) cortical granule exocytosis and (5) maternal mRNA replication. This complex process needs a control in the homeostasis that will be basically regulated by the mitochondria present in the oocyte and by some channels in the ER and the oolemma. Oocyte activation failure is caused by the absence of Ca2+ oscillations or by alteration of their frequency. This failure can be related with both gametes. Regarding sperm, deficiencies in PLCζ have been reported to be directly associated with the absence of Ca2+ oscillations. As for oocytes, and even though studies are scarce due to ethical restrictions, certain hypotheses have proposed activation failure to be associated with deficiencies in mechanisms of homeostasis. This actually means failures in mitochondria and related channels, and also with some molecules like PIP2, DAG, PLC isoforms and IP3R. In order to avoid oocyte activation failure, assisted reproduction clinics have developed certain protocols for assisted oocyte activation, being those with calcium ionophores the most widely extended in fertility centres. REFERENCES
Authors: Javier del Río, Belén Gómez-Giménez & Iris Martínez "The relationship between blastocyst morphology and implantation has been investigated mainly according to expansion, trophectoderm and inner cell mass"  Figure 1. Human blastocysts (10). The main goal of in vitro fertilization (IVF) is the birth of a single healthy child. However, the consequences and the effects of assisted reproductive techniques on children’s short- and long-term health have always been a source of discussion. Although IVF techniques and protocols have dramatically improved, the overall success rates are still relatively low, and assisted reproduction units still face the challenge of improving pregnancy rates (1). For this purpose, transfer of a single human embryo at blastocyst stage is becoming more common in the practice of assisted reproduction (2). It allows a better synchronization between the endometrium and the embryo and the possible selection of embryos with a higher implantation potential (3). Several morphology- or kinetics-based approaches have been described to select the best blastocyst in order to increase pregnancy rates. However, the yielded results are conflicting and the outcome is a matter of never-ending and controversial debates, specially regarding blastocyst stage (4, 5). BLASTOCYST ASSESSMENT SYSTEMS The relationship between blastocyst morphology and subsequent blastocyst implantation has been investigated according to various criteria. Traditionally, morphology has been evaluated after embryo compaction (6). The significance of examining the embryo after compaction is the ability to examine it after embryonic genome activation. Furthermore, the obvious benefit of looking at the blastocyst is the possibility to examine both cell types. The extent to which the trophectoderm (TE) develops will reflect the embryo’s ability to attach and implant in the endometrium, whereas development of the inner-cell mass (ICM) is obviously crucial for the progress of the foetus (7). There have been described several assessment systems to predict the success of blastocyst implantation. However, Gardner's grading system seems to be a better predictor of pregnancy rates (8, 4, 7). Following this method, blastocysts are initially scored from 1 to 6 based on their degree of expansion and hatching status, and ICM and TE grading is then assessed from A to D depending on their morphology (9). It was felt that expansion was important for cavity formation. This process requires both extensive energy utilization through the Na+/K+ ATPases on the basolateral membrane of the TE and formation of effective tight junctions between TE cells to form a barrier. Therefore, expansion seems to be a reflection of embryo competence (7). Recently, Richardson et al. proposed a simplified blastocyst grading system. These authors demonstrated both its prognostic potential and the inter- and intra-observer variability. This grading scheme was able to effectively predict clinical outcomes in terms of implantation, clinical pregnancy and live birth. Slight variation existed both between and within embryologists grading the embryos but, overall, consistency in their analyses was similar to, if not better than, those associated with more complex grading systems (10). However, most of the grading systems that are currently used for assessing viability of IVF embryos are subjective, relying on visual inspection of morphological characteristics of the embryos that are qualitatively evaluated. Grading based on qualitative criteria is imprecise, and it inevitably results in inter-observer variability and in intra-observer to some extent, as well (10). MORPHOLOGICAL ASSESSMENT AND OUTCOME RESULTS As it has been exposed, there is a need for increased knowledge about the relative impact of each morphology parameter at the blastocyst stage (and their potential correlation) on predicting the probability of successful implantation and pregnancy (1, 2, 11). Shapiro et al. compared up to 25 parameters in order to develop predictive models of clinical pregnancy within a set of blastocyst transfer cycles (12). Among these variables, blastocyst diameter seemed to be the most significant predictor of clinical pregnancy in the multivariate models. The authors concluded that embryos developing into expanded blastocyst stage on day 5 were approximately twice as likely to implant, compared to those for which expansion was delayed until day 6 (13, 3). This is supported by Van den Abbeel and coauthors, who found that high scores of blastocyst expansion and hatching stage, ICM and TE grade were all significantly associated with increased pregnancy and live birth rates after fresh transfers (11). The finding that the expansion and hatching stage is the most important parameter when selecting a blastocyst for transfer (11) is in contrast with some retrospective cohort studies that suggest TE grading to have the strongest predictive power for treatment outcome in fresh transfers (14, 15). On the contrary, Basak Balaban et al. exposed that quantitative measurement of blastocysts and ICM is not a practical way to assess blastocyst quality, arguing that two-dimensional measurements of three-dimensional global structures can be misleading. The reasoning is that the size of a blastocyst may vary depending on the time the blastocyst is assessed under the microscope, and this may easily confuse grading (9). For this purpose, Almagor et al. tried to provide an easily measurable assessment of the ICM and evaluate its correlation with pregnancy rates in a series of single blastocyst transfers. They found a high ICM/blastocyst ratio associated with significantly increased pregnancy rates. Thus, they proposed this measure to be used as an additional strongly predictive parameter of successful implantation (16). Recently, Bouillon et al. have confirmed that clinical pregnancy and live birth rates were significantly higher for blastocysts with good TE and ICM quality, and so it was concluded that both rates decreased with morphology (4). Even though some blastocysts with non-optimal morphology are able to implant, it has been suggested that when selection is made among suboptimal blastocysts, preference should be given to those with a normal ICM (6). However, the current goal for researchers is to establish the optimal perinatal outcome of singletons according to blastocyst morphology. This has been recently analyzed by Bouillon et al., who found no increased rates of adverse obstetric and perinatal outcomes after transfer of blastocysts with poor morphological features (4).  Figure 2. Examples of blastocyst grading: (a) 3AA blastocyst; (b) 3AB blastocyst; (c) 3BA blastocyst; (d) 4AA blastocyst; (e) 4AB blastocyst; (f) 4BA blastocyst; (g) 4CC blastocyst; (h) 5AA blastocyst; (i) 5CA blastocyst. For details of the EH stages and ICM and TE grades, see Materials and methods from Van den Abbeel (11). BLASTOCYST ASSESSMENT BY TIME-LAPSE TECHNOLOGY As previously explained, the most accepted blastocyst grading system is Gardner’s (17), based on the degree of blastocyst expansion and the morphological appearance of both the ICM and TE. However, since embryo development is a dynamic process, conventional grading practices may not detect subtle differences in morphology, which changes significantly over a time span of only a few hours (18). In order to obtain a complete picture of morpho-kinetic events occurring during embryo development a time-lapse system is needed. This technology offers continuous monitoring of embryos rather than just a limited number of discrete observations annotated through conventional assessment. Besides, time-lapse allows embryos to be cultured uninterruptedly, thus getting rid of embryo trafficking from and into the incubator (19). Nevertheless, the actual new and unique contribution of morpho-kinetics is the ability to predict how likely is for a zygote to reach the blastocyst stage in vitro. Several algorithms based on parameters detected by time-lapse, such as early divisions of cleavage-stage embryo, have recently been developed in IVF laboratories to predict blastocyst formation (20). In addition, some authors have made an effort to take time-lapse usefulness further, for instance, to predict the ploidy status of pre-implantation embryos (21, 22). Implantation potential of blastocysts can be evaluated by means of time-lapse during its development. In this regard, three main events are currently being investigated: duration of both compaction and blastulation plus number of blastocyst collapse events (19, 23, 24). Duration of compaction After several cell divisions during the initial stages of embryonic development, the intercellular boundaries become obscured in a process called compaction, which maximizes the intercellular contact and gives rise to the morula (25). Although the compaction of embryos has not received sufficient attention in the IVF field, some studies have focused on the relationships between compaction patterns and embryo developmental potential. Embryos that begin to compact before the eight-cell stage exhibit aberrant in vitro development. Conversely, embryos that complete compaction on day 5 have a lower ability to develop into high-quality blastocysts than those that compact on day 4 (26). These results suggest that the compaction patterns of embryos can facilitate the prediction of their ability to develop both in vitro and in vivo. An interesting work on this issue has been recently published by Mizobe and collaborators (23). The study retrospectively examined the outcome of 299 embryos from 243 patients, which were transferred at blastocyst stage. The whole early development was analysed by comparing morpho-kinetic parameters between implanted and non-implanted embryos, and measuring the time length of specific events, particularly of embryo compaction. Compaction length was calculated by using values of beginning and end of compaction. Beginning of compaction was considered as the time point when the intercellular boundaries became diffuse somewhere in the embryo, while fully compaction was defined as the point when blastomeres were finally unified into one cluster. Compaction length was significantly shorter in blastocysts resulting in pregnancies compared to those that failed to do so. These results indicate a correlation between the length of compaction and implantation potential. This finding is in agreement with the results from previous studies, which observed that the compaction patterns of embryos affected the rates of good-quality blastocyst formation and implantation (26, 27, 28). By contrast, some studies have reported that compaction time of embryos does not affect clinical pregnancy rates (29, 30). 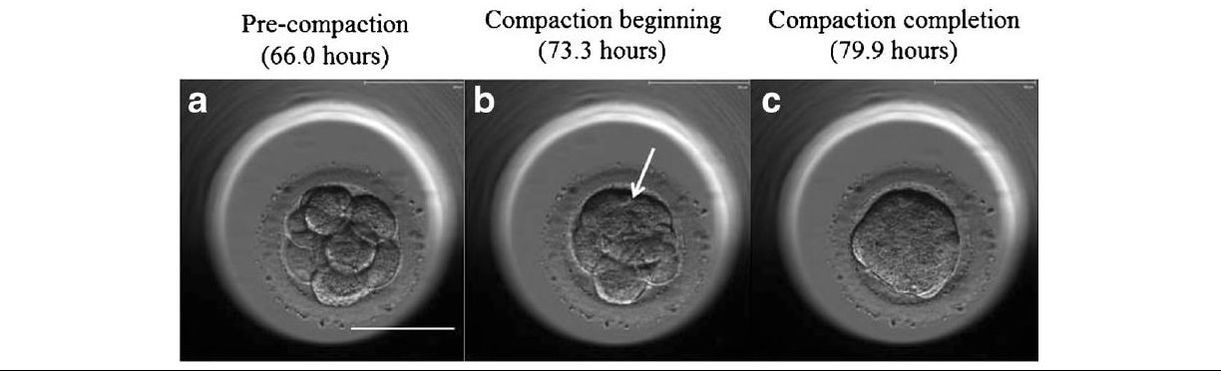 Figure 3. Optimal compaction timing of a blastocyst according to Mizobe and colleagues (23). Duration of blastulation Blastulation is the process through which a morula becomes a blastocyst. Two different structures will arise to form the blastocyst out of the compacted blastomeres of the morula. The first sign of blastulation is compaction and differentiation of the outer blastomeres, forming the TE. This compaction gives the structure a watertight condition, allowing the fluid later secreted to be contained (31). Then, a different group of blastomeres normally located at the centre of the morula start to get closely attached to each other by the formation of Gap junctions, thus facilitating cell communication. It is these cells that differentiate into the ICM (the future embryoblast) and acquire a polarized location at one edge of the embryo. Such polarization creates a cavity, the blastocoel, and gives rise to the structure termed blastocyst. The trophoblasts (TE cells), in turn, continuously pump fluid into the blastocoel, which results in an enhanced size of the blastocyst. This increased volume leads the embryo to hatch through the zona pellucida (32). A recent study conducted by Mumusoglu analysed whether time-lapse morpho-kinetic variables differ among those euploid blastocysts that result in ongoing pregnancy after single embryo transfer (24). For that purpose, 129 patients who had been transferred a single embryo after an ICSI cycle with PGS were considered. Embryos were cultured in a time-lapse incubator up to the moment of TE biopsy, and 23 time-lapse morpho-kinetic parameters were annotated. After biopsy, blastocysts were vitrified and transferred within the next cycle. When comparing all time-lapse parameters, only blastulation time was statistically different: it had lasted shorter in successfully implanted blastocysts than in those that had not implanted. Blastulation time was calculated as the interval from initiation of blastulation up to full blastocyst formation (33, 34). Even though only a few studies have genetically tested euploid blastocysts (21, 22), all of them have pointed out that faster-developing euploid blastocysts might exhibit higher implantation potential. Even so, further large-scale studies are needed in order to confirm such an association (24). Blastocyst collapse events The phenomenon of blastocyst collapse is actually the shrinkage caused by the efflux of the blastocoel fluid due to the loss of cell bindings along the TE. When blastocysts expand, fluid gradually accumulates in the blastocoel -mediated by the sodium pump (Na+/K+-ATPase) (35), resulting in an increased pressure on both the TE and the zona. In parallel, TE cells produce lysins that are involved in the zona weakening and hatching. Formerly to implantation, the embryo needs to leave the zona behind, place adjacent to the endometrial epithelium and then make first contact with the uterus (36). Thus, embryo hatching from the zona is thought to be related to collapse-expansion cycles. By using a time-lapse monitoring system, it has been observed that many of the human blastocysts that reach stage 5 of expansion experience one or more collapse events of the blastocoel cavity, producing a separation of part (if not all) of the TE cells from the zona (19). In a study conducted by IVI Valencia and IVI Murcia clinics (19), blastocyst collapse was analysed to determine its potential influence on reproductive outcomes and whether it may serve for prognostic purposes. 460 patients and data from over 500 blastocysts known to have implanted were included in the study. Blastocyst collapse was considered to have occurred if the separation between TE and the zona pellucida was higher than 50% of the volume. Blastocysts that had experienced just one collapse event were found to present a significantly reduced implantation potential when compared to those transferred after having experienced none. The authors proposed that the molecular mechanisms underlying this association could be related to the mechanical stress suffered from by the embryo, which could result in an excessive energy consumption that would adversely affect the consequent development (19).  Figure 4. Drawing tools used with Embryovieverw for blastocyst collapse evaluation. First, a line was drawn across the embryo diameter (A). Then, the two circumferences that define the contracted blastocyst and the inner surface of the zona pellucida were outlined (B) [for more details, go to Materials and methods from Marcos (19)]. In spite of the data discussed above, the negative association between blastocyst collapse and implantation potential is not yet clear. In a report by Bodri and colleagues (37), blastocysts were classified according to the number of collapses: embryos with no collapses represented 54% of the total, 22% of the embryos had experienced one single collapse, and multiple collapse events occurred in 24% of the blastocysts. Whereas the live birth rate was observed to decrease as the number of embryo collapse increased, multivariate analyses suggested blastocyst collapse not to be a significant predictor. Rather, it was found to be a confounding factor, along with other morpho-kinetic variables such as time up to two-cell division completion and female age. Therefore, it was concluded that blastocyst collapse patterns should not be evaluated alone without stronger predictors of reproductive outcomes being taken into account (37). TIME-LAPSE AS A MEANS TO EVALUATE EMBRYO QUALITY As previously exposed, the use of time-lapse technology is recently common in embryology laboratories because of its noticeable potential for enhancing embryo selection. Using these technologies, Desai et al. analysed possible kinetic differences between embryos with limited potential and those that accomplished in vitro blastocyst formation and/or implantation (38). Certain parameters such as time of pronuclear formation and cleavage stage were found to be different in embryos reaching blastocyst stage vs. poor-quality embryos. Moreover, a large number of embryos were found to present multinucleation and reverse cleavage, but they were able to form a blastocyst with optimal criteria for freezing (38), which resembles previous reports on the dynamic nuclear formation of blastocysts by Ergin and coauthors (39). With respect to blastocyst formation, Motato et al. (2016) proposed two models to classify embryos based on their probability of reaching blastocyst stage and implantation (40). However, the study was limited by parameters such as subjective criteria from different clinics with different culture media (40). Consequently, it would be reasonable to keep on research on this subject in order to achieve a consensus regarding embryo classification and implantation potential (40). CONSIDERING DAY-2 AND DAY-3 EMBRYO MORPHOLOGY BEFORE DAY-5 TRANSFER Even though blastocyst stage is currently widely accepted as the optimal moment for embryo transfer, cleavage stage has been traditionally regarded as the right moment in global practice. In fact, it still continues to be so in some laboratories, and early transfer into the uterus has been proposed to be advantageous to the embryo due to the limited time exposed to the in vitro environment (41). However, there exist two main arguments supported by extensive scientific literature to explain why blastocyst transfer after extended culture has advantages over the traditional cleavage-stage transfer: First of all, when the embryo arrives to the uterus in natural conditions it has already reached morula stage, which corresponds to, at least, day 4 of in vitro culture (42). This means blastocyst stage is the most physiologically compatible stage for transfer, since it allows a better synchronization between embryonic stage and endometrial receptivity (43) [you can read more about the optimal day for embryo transfer in our previous article here]. Secondly, several studies have reported higher implantation potential for blastocysts compared to cleavage-stage embryos (6, 41), the first transferred blastocyst being reported in 1995 (44). Furthermore, some authors have postulated that a large proportion of morphologically normal day-3 embryos are actually chromosomally abnormal or mosaic, which may contribute to the 80-90% rate of implantation failure observed after cleavage-stage embryo transfer (45). EXTENDING EMBRYO CULTURE UP TO BLASTOCYST STAGE Considering the need for further studies on the subject, and the fact that day-3 embryos can actually implant and develop successfully, does it really make sense to extend embryo culture up to blastocyst stage? As above-stated, morphologically normal embryos may actually present chromosome abnormalities, which proves the insufficiency of morphological criteria to evaluate implantation rates (46). Because of embryo plasticity, the proportion of chromosomally abnormal cells varies within the culture; corrupted cells can be eliminated, thus resulting in a good-quality blastocyst developing from a poor-quality cleavage-stage embryo (47). Some studies have evaluated pregnancy rates derived from transfers of blastocysts with previous poor quality as cleavage-stage embryos, finding an approximate success rate of 45% after culture and freezing of embryos at an early stage for another cycle. The conclusion of this being a valid practice to avoid the repetition of IVF-ICSI treatments (48) agrees with recent findings showing that low-scoring day-2 or 3 embryos, which are not considered transferable, can still result in successful blastulation and end up in a live birth (49, 50). All this said, the right question now would be: should day-2 and day-3 embryo morphology be considered before transfer at day 5 when blastocysts reach a similar good quality? A recent retrospective study by Herbemont has suggested that only the quality of the transferred blastocyst may be predictive of the subsequent clinical outcome, whereas morphological aspects at day 2 or day 3 have limited interest (51). These same results had previously been observed by Guerif; even though early morphological parameters were relatively helpful to predict blastocyst development, their value to predict blastocyst morphology was limited, and so they provided no significant additional information that could prognosticate blastocyst implantation and live birth rates (6). A few years earlier and with the same goal in mind, Zech and coauthors carried out a prospective randomized study in which they compared ongoing pregnancy rates per single embryo transfer between day 3 and day 5. When good-quality embryos were available, pregnancy rates were found to be higher after blastocyst transfer. Therefore, the authors concluded that morphological criteria-based seleccion at day 3 may not be a suitable procedure when just one embryo is to be transferred out of a cohort of all morphologically good ones. Thus, and as stated by the authors, extending embryo culture up to day 5 may result in a better strategy in order to correctly identify and select those embryos with higher implantation potential, provided there is a sufficient number of top-quality eight-cell embryos available (52). On the contrary, a study performed by Silber (2014) found that blastocysts arising from poor-quality embryos displayed lower implantation and pregnancy rates compared to good-quality embryos. These discrepancies could be due to different criteria used to score embryo quality (53). So, in order to minimise discrepancies between studies, the use of time-lapse is currently established as a common approach to evaluate embryo morpho-kinetics. In fact, reduction in the time of embryo exposure to the environment outside the incubator has been demonstrated to enhance both embryo quality and blastulation rates (54). Nevertheless, to answer the question previously postulated, a prospective randomized study would be needed that compare at least two similar good-quality blastocysts, one arising from a good-quality day-2/3 embryo, and to the other from a poor-quality one (51). CONCLUSIONS It is important to take into account that the main population features of different patients, such as paternal age, maternal BMI, parental smoking or cause of infertility may influence clinical outcomes. Moreover, certain methodological aspects also need to be considered, like blastocyst evaluation by the same personal (in order to minimize variation) or the consistent use of the same type of culture media (to avoid potential effects on birth-weight and other traits), just as previously suggested (4). A universal embryo grading system needs to be validated, before widespread implementation in IVF laboratories. Also, it has not yet been clearly established which morphological feature of blastocysts (expansion, TE or ICM) is the most reliable as a predictive factor for post-transfer implantation success. Consequently, there is still a debate between authors about the true outcomes of single transfer of low-quality blastocysts (4). Morpho-kinetics assessment, along with chromosomal screening, may ultimately help identify euploid embryos with the highest developmental potential (55). Since these features are susceptible of being affected by in vitro culture conditions, each embryology laboratory should define their own cut-off points in order to standardise time-lapse variables (24). Finally, it should be taken into account the fact that embryo quality is not the only parameter with influence on implantation rates; endometrial receptivity is also involved, and it may be greatly determined by a variety of factors (56) [to learn more about endometrium status and receptivity, read our previous post here]. REFERENCES
1- Souza Setti A, Ferreira RC, Paes de Almeida, Ferreira Braga D, de Cassia Savio Figueira R, Iaconelli A Jr, Borges E Jr. Intracytoplasmic sperm injection outcome versus intracytoplasmic morphologically selected sperm injection outcome: a meta-analysis. Reprod Biomed Online 2010;21:450-455. 2- Meldrum DR. Blastocyst transfer: a natural evolution. Fertil Steril 1999;72:216–7. 3- Balaban B, Urman B, Sertac A, Alatas C, Aksoy S, Mercan R. Blastocyst quality affects the success of blastocyst-stage embryo transfer. Fertil Steril 2000;74:28-287. 4- Bouillon C, Celton N, Kassem S, Frapsauce C, Guérif F. Obstetric and perinatal outcomes of singletons after single blastocyst transfer: is there any difference according to blastocyst morphology? Reprod Biomed Online 2017;35:197–207. [Internet]. 5- Stecher A, Vanderzwalmen P, Zintz M, Wirleitner B, Schuff M, Spitzer D, Zech NH. Transfer of blastocysts with deviant morphological and morphokinetic parameters at early stages of in-vitro development: a case series. Reprod Biomed Online 2014;28:424-435. 6- Guerif F, Lemseffer M, Leger J, Bidault R, Cadoret V, Chavez C, Gasnier O, Saussereau MH, Royere D. Does early morphology provide additional selection power to blastocyst selection for transfer? Reprod Biomed Online 2010;21:510– 519. 7- Alpha Scientists in Reproductive Medicine and ESHRE Special Interest Group of Embryology. The Istanbul consensus workshop on embryo assessment: proceedings of an expert meeting. Hum Reprod 2011;26:1270 –1283. 8- Gardner DK, Schoolcraft WB, Wagley L, Schlenker T, Stevens J, Hesla J. A prospective randomized trial of blastocyst culture and transfer in in-vitro fertilization. Hum Reprod 1998;13:3434-3440. 9- Balaban B, Yakin K, Urman B. Randomized comparison of two different blastocyst grading systems. Fertil Steril 2006;85:559– 563. 10- Richardson A, Brearley S, Ahitan S, Chamberlain S, Davey T, Zujovic L, Hopkisson J, Campbell B, Raine-Fenning N. A clinically useful simplified blastocyst grading system. Reprod Biomed Online 2015;31:523–530. 11- Van den Abbeel E, Balaban B, Ziebe S, Lundin K, Cuesta MJ, Klein BM, et al. Association between blastocyst morphology and outcome of singleblastocyst transfer. Reprod Biomed Online 2013;27:353–61. 12- Shapiro BS, Daneshmand ST, Garner FC, Aguirre M, Thomas S. Large blastocyst diameter, early blastulation, and low preovulatory serum progesterone are dominant predictors of clinical pregnancy in fresh autologous cycles. Fertil Steril 2008;90:302–9. 13- Shapiro BS, Richter KS, Harris DC, Daneshmand ST. A comparison of day 5 and 6 blastocyst transfers. Fertil Steril 2001;75:1126–30. 14- Ahlström A, Westin C, Reismer E, Wikland M, Hardarson T. Trophectoderm morphology: an important parameter for predicting live birth after single blastocyst transfer. Hum Reprod 2011;26:3289– 3296. 15- Hill MJ, Richter KS, Heitmann RJ, Graham JR, Tucker MJ, Decherney AH, Browne PE, Levens ED. Trophectoderm grade predicts outcomes of single-blastocyst transfers. Fertil Steril 2013;99: 1283–1289. 16- Almagor M, Harir Y, Fieldust S, Or Y, Shoham Z. Ratio between inner cell mass diameter and blastocyst diameter is correlated with successful pregnancy outcomes of single blastocyst transfers. Fertil Steril 2016;106: 1386–1391. [Internet] 17- Gardner DK, Lane M, Stevens J, Schlenker T, Schoolcraft WB. Blastocyst score affects implantation and pregnancy outcome: towards a single blastocyst transfer. Fertil Steril. 2000;73(6):1155–8. 18- Cruz M, Garrido N, Herrero J, Pérez-Cano I, Muñoz M, Meseguer M. Timing of cell division in human cleavage-stage embryos is linked with blastocyst formation and quality. Reprod Biomed Online. 2012;25(4):371–81. 19- Marcos J, Pérez-Albalá S, Mifsud A, Molla M, Landeras J, Meseguer M. Collapse of blastocysts is strongly related to lower implantation success: a time-lapse study. Hum Reprod. 2015;30(11):2501–8. 20- Meseguer M, Herrero J, Tejera A, Hilligsøe KM, Ramsing NB, Remohí J. The use of morphokinetics as a predictor of embryo implantation. Hum Reprod. 2011;26(10):2658–71. 21- Yang Z, Zhang J, Salem SA, Liu X, Kuang Y, Salem RD, et al. Selection of competent blastocysts for transfer by combining time-lapse monitoring and array CGH testing for patients undergoing preimplantation genetic screening: a prospective study with sibling oocytes. BMC Med Genomics. 2014;7(1):38. 22- Kofinas JD, Tiegs A, Kramer YG, McCulloh DH, Grifo JA. Do Time Lapse Morphokinetic (TLM) Parameters Distinguish between Good Versus Poor Prognosis Embryos of Known Ploidy Status? Fertil Steril. 2015;103(2):e27. 23- Mizobe Y, Ezono Y, Tokunaga M, Oya N, Iwakiri R. Selection of human blastocysts with a high implantation potential based on timely compaction. J Assist Reprod Genet. 2017;34(8):991-997. 24-Mumusoglu S, Ozbek IY, Sokmensuer LK, Polat M, Bozdag G, Papanikolaou E, et al. Duration of blastulation may be associated with ongoing pregnancy rate in single euploid blastocyst transfer cycles. Reprod Biomed Online. 2017;35(6):633-639. 25- Fleming TP, Sheth B, Fesenko I. Cell adhesion in the preimplantation mammalian embryo and its role in trophectoderm differentiation and blastocyst morphogenesis. Front Biosci. 2001;6(1):D1000–7. 26- Iwata K, Yumoto K, Sugishima M, Mizoguchi C, Kai Y, Iba Y, et al. Analysis of compaction initiation in human embryos by using time-lapse cinematography. J Assist Reprod Genet. 2014;31(4):421–6. 27- Skiadas CC, Jackson K V, Racowsky C. Early compaction on day 3 may be associated with increased implantation potential. Fertil Steril. 2006;86(5):1386–91. 28- Tao J, Tamis R, Fink K, Williams B, Nelson-White T, Craig R. The neglected morula/compact stage embryo transfer. Hum Reprod. 2002;17(6):1513–8. 29- Chamayou S, Patrizio P, Storaci G, Tomaselli V, Alecci C, Ragolia C, et al. The use of morphokinetic parameters to select all embryos with full capacity to implant. J Assist Reprod Genet. 2013;30(5):703–10. 30-Kirkegaard K, Kesmodel US, Hindkjær JJ, Ingerslev HJ. Time-lapse parameters as predictors of blastocyst development and pregnancy outcome in embryos from good prognosis patients: a prospective cohort study. Hum Reprod. 2013;28(10):2643–51. 31- Forgacs G, Newman SA. Biological physics of the developing embryo. Cambridge University Press; 2005. 32- Larsen WJ. Human embryology. Churchill Livingstone; 2001. 33- Campbell A, Fishel S, Bowman N, Duffy S, Sedler M, Hickman CFL. Modelling a risk classification of aneuploidy in human embryos using non-invasive morphokinetics. Reprod Biomed Online. 2013;26(5):477–85. 34- Ciray HN, Campbell A, Agerholm IE, Aguilar J, Chamayou S, Esbert M, et al. Proposed guidelines on the nomenclature and annotation of dynamic human embryo monitoring by a time-lapse user group. Hum Reprod. 2014;29(12):2650–60. 35- Baltz JM, Smith SS, Biggers JD, Lechene C. Intracellular ion concentrations and their maintenance by Na+/K+-ATPase in preimplantation mouse embryos. Zygote. 1997;5(1):1–9. 36- Biggers JD. Reflections on the culture of the preimplantation embryo. Int J Dev Biol. 2004;42(7):879–84. 37- Bodri D, Sugimoto T, Serna JY, Kawachiya S, Kato R, Matsumoto T. Blastocyst collapse is not an independent predictor of reduced live birth: a time-lapse study. Fertil Steril. 2016;105(6):1476–83. 38-Desai N, Ploskonka S, Goodman LR, Austin C, Goldberg J, Falcone T. Analysis of embryo morphokinetics, multinucleation and cleavage anomalies using continuous time-lapse monitoring in blastocyst transfer cycles. Reprod Biol Endocrinol. 2014;12:54. 39- Ergin EG, Calişkan E, Yalçinkaya E, Oztel Z, Cökelez K, Ozay A, et al. Frequency of embryo multinucleation detected by time-lapse system and its impact on pregnancy outcome. Fertil Steril. 2014;102(4):1029-33.e1. 40- Motato Y, de los Santos MJ, Escriba MJ, Ruiz BA, Remohí J, Meseguer M. Morphokinetic analysis and embryonic prediction for blastocyst formation through an integrated time-lapse system. Fertil Steril. 2016;105(2):376-84.e9. 41- Glujovsky D, Farquhar C, Quinteiro Retamar AM, Alvarez Sedo CR, Blake D. Cleavage stage versus blastocyst stage embryo transfer in assisted reproductive technology. Cochrane Database Syst Rev. 2016(6):CD002118. 42- Croxatto HB, Fuentealba B, Díaz S, Pastene L, Tatum HJ. A simple nonsurgical technique to obtain unimplanted eggs from human uteri. Am J Obstet Gynecol. 1972;112(5):662-8. 43- Valbuena D, Martin J, de Pablo JL, Remohí J, Pellicer A, Simón C. Increasing levels of estradiol are deleterious to embryonic implantation because they directly affect the embryo. Fertil Steril. 2001;76(5):962-8. 44- Edwards RG, Brody SA. History and ethics of assisted human conception. Principles and Practice of Assisted Human Reproduction. Philadelphia: WB Sauders, 1995:17–47. 45- Magli MC, Gianaroli L, Munné S, Ferraretti AP. Incidence of chromosomal abnormalities from a morphologically normal cohort of embryos in poor-prognosis patients. J Assist Reprod Genet. 1998;15(5):297-301. 46- Meseguer M, Rubio I, Cruz M, Basile N, Marcos J, Requena A. Embryo incubation and selection in a time-lapse monitoring system improves pregnancy outcome compared with a standard incubator: a retrospective cohort study. Fertil Steril. 2012;98(6):1481-9.e10. 47- Fragouli E, Alfarawati S, Spath K, Jaroudi S, Sarasa J, Enciso M, et al. The origin and impact of embryonic aneuploidy. Hum Genet. 2013;132(9):1001-13. 48- Kaartinen N, Das P, Kananen K, Huhtala H, Tinkanen H. Can repeated IVF-ICSI-cycles be avoided by using blastocysts developing from poor-quality cleavage stage embryos? Reprod Biomed Online. 2015;30(3):241-7. 49- Hershko Klement A, Ovadia M, Wiser A, Berkovitz A, Shavit T, Nemerovsky L, et al. What we learned from extended culture of 'rejected' day-3 cleavage stage embryos: a prospective cohort study. J Ovarian Res. 2017;10(1):35. 50- Sallem A, Santulli P, Barraud-Lange V, Le Foll N, Ferreux L, Maignien C, et al. Extended culture of poor-quality supernumerary embryos improves ART outcomes. J Assist Reprod Genet. 2017. 51- Herbemont C, Sarandi S, Boujenah J, Cedrin-Durnerin I, Sermondade N, Vivot A, et al. Should we consider day-2 and day-3 embryo morphology before day-5 transfer when blastocysts reach a similar good quality? Reprod Biomed Online. 2017;35(5):521-8. 52- Zech NH, Lejeune B, Puissant F, Vanderzwalmen S, Zech H, Vanderzwalmen P. Prospective evaluation of the optimal time for selecting a single embryo for transfer: day 3 versus day 5. Fertil Steril. 2007;88(1):244-6. 53- Silber, S., 2014. Blastocysts that came from better quality day 3 embryos give higher pregnancy rates than blastocysts that derive from poor quality day 3 embryos. Ovarian Club, Paris. 54- Zhang JQ, Li XL, Peng Y, Guo X, Heng BC, Tong GQ. Reduction in exposure of human embryos outside the incubator enhances embryo quality and blastulation rate. Reprod Biomed Online. 2010;20(4):510-5. 55- Zaninovic N, Irani M, Meseguer M. Assessment of embryo morphology and developmental dynamics by time-lapse microscopy: is there a relation to implantation and ploidy? Fertil Steril. 2017;108(5):722-9. 56- Valbuena D, Valdes CT, Simon C. Introduction: Endometrial function: facts, urban legends, and an eye to the future. Fertil Steril. 2017;108(1):4-8. Authors: Iñaki Arroyos, María Caballero & Raquel Pillado "Up to date, clinical practice has proven that the use of in vitro culture for human embryos does not imply any major adverse consequences on the offspring" 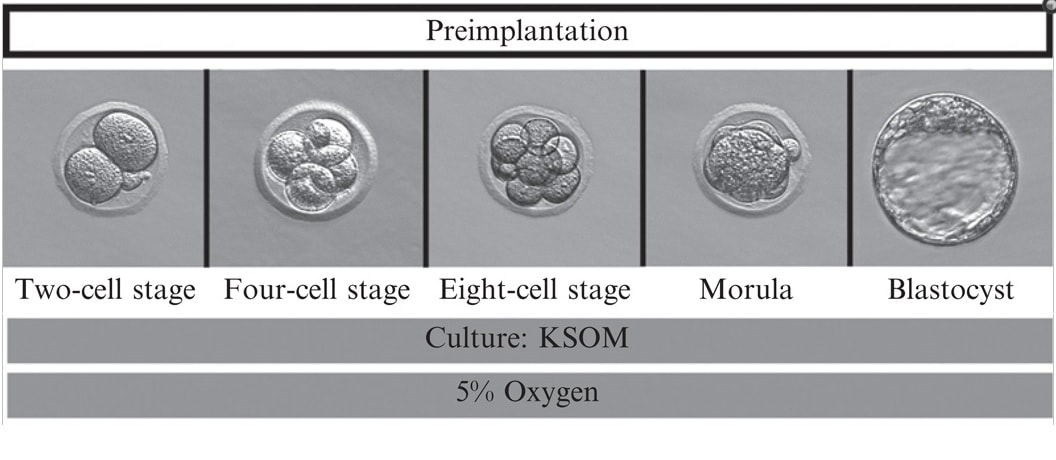 Figure 1. Mouse embryo culture from 2-cell stage to blastocyst using KSOM medium (1). Figure 1. Mouse embryo culture from 2-cell stage to blastocyst using KSOM medium (1). INTRODUCTION - Why is it important to talk about IVF culture media? Within the last 40 years, the improvement of assisted reproduction technologies (ART) has experienced rapid and great advances (2). This success is largely due to the important enhancement of the culture conditions, specially IVF media (2,3), since this is where the early embryo develops up to blastocyst stage (Figure 1) before being transferred into the womb. However, it is currently known that in spite of this huge improvement regarding culture media, there still exist certain important associated-risks. These include foetal growth restriction, premature birth, low birth weight, congenital anomalies, perinatal complications and even epigenetic alterations (2,3,4). Therefore, there is still a need to improve our current knowledge on IVF media components, concentrations and related factors, in order to dodge those potential negative effects. - How did culture media evolve through history? The development of culture media for human embryos has been possible thanks to many years of animal research (Figure 2). The first mouse embryo culture took place in plasma (5) and blood clot (6). Later on, development of synthetic media and simple culture media began, with improvements such as salt solutions supplemented with glucose, egg white and yolk (7) or Krebs-Ringer-bicarbonate (KRM) solution with glucose, bovine serum albumin (BSA) and antibiotics (8). However, embryos used at that time were collected from the Fallopian tubes at the 8-cell stage, thus being impossible to address development from the very 1-cell stage (2). Elements such as the appropriate pH level and osmotic pressure, the effect of energy substrates, BSA and amino acids in the embryo culture were widely studied during the 60’s. Glucose was replaced by lactate and pyruvate upon Brinster's demonstration that the 2-cell-stage embryo did not use glucose as energy source, and that its presence at certain concentrations hampered the 2-cell embryo development to blastocyst (10). The first success in human IVF was achieved by Edwards and Steptoe in 1969, who used a medium based on a modification of Tyrode’s solution (10). Two years later, using a more complex medium called Ham’s F-10 (made of inorganic salts, glucose, pyruvate, amino acids, vitamins and serum, among others), Steptoe and collaborators succeeded in culturing human embryos from the zygote to blastocyst (11). In 1978 and using this medium, Edwards and Steptoe got the first embryo transfer that resulted in the birth of the first IVF baby, Louise Brown (12). However, over the years, some elements from Ham's F-10 medium were shown to have adverse effects on embryo development, and so its usage was stopped (13). Years passed and different modifications took place: the removal of proteins from the media, the use of amniotic fluid and, finally, the search of the “optimal” medium. For this approach, around 1970 the development of the following media was based on the “back to nature” vision (2). New media were developed according to the composition of the reproductive (tubal and uterine) fluids (as an example, the HTF medium was made only of inorganic salts, glucose, pyruvate, lactate, human serum albumin and antibiotics). Nevertheless, glucose and phosphate were reported to be toxic to cleavage-stage embryos (as mentioned above), glutamine replaced the glucose and ethylenediaminetetraacetic acid (EDTA) was added instead of phosphate. Blastocyst formation rates were not entirely satisfactory; subsequent studies found that amino acids and vitamins improved these rates because the toxic effect of glucose was avoided (reviewed in 13). At the same time, sophisticated software was used to get a simplex optimization medium by getting the concentration of each medium component in vitro. Using mouse embryo assays, the potassium simplex optimized medium (KSMO) (14) and the KSOMAA medium (KSMO supplemented with amino acids (15)) were developed. These media are effective for the human embryo culture and, in fact, KSOMAA is widely used in human ART under the brand of Global (13). 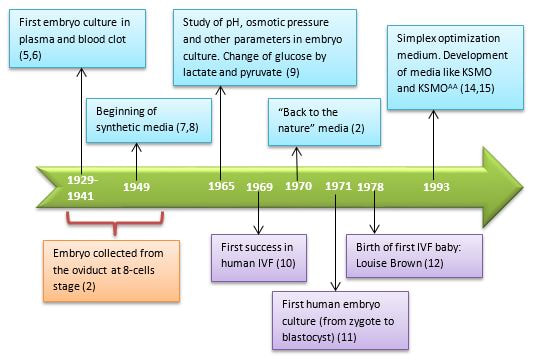 Figure 2. Overview of culture media development through history. WHICH MEDIA DO WE USE NOW? - Components Nowadays, the market of IVF culture media is widely expanded worldwide. Cook Medical, Cooper Surgical (producer of both SAGE and ORIGIO media), FertiPro, Gynotec, Genea Biomedx, InVitroCare and Kitazato are some of the companies that produce culture media for human IVF (2). Even though all commercial media present the same components (see Table 1) (2), all of them well known by every company, the concentrations and the most basic parameters for embryo culture such as the use of amino acids and glucose, temperature or gas composition, vary between brands (16). 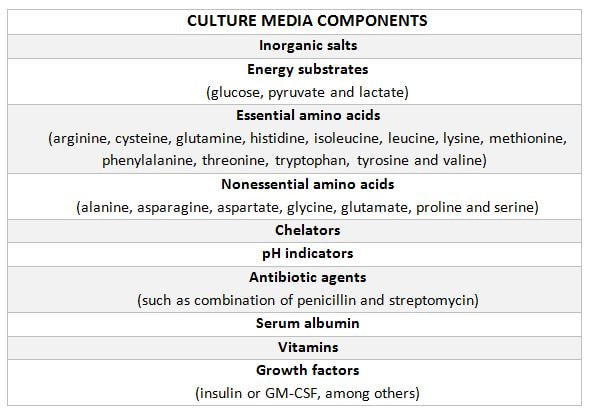 Table 1. Composition of current comercial culture media (2). - Different embryo needs (time/development stage) It is important to bear in mind the nutritional requirements of the embryo during development. Several authors have found that the required energy substrates, amino acids and proteins concentrations differed according to the embryo stage. Based on these results, it was established that between days 1 and 3 (cleavage stage), glucose should be reduced or removed if there are no amino acids or EDTA in the medium, in order to avoid the adverse effects of glucose. Lactate, pyruvate, taurine and non-essential amino acids should be included as well, but essential amino acids should be omitted at this point. A protein source such as pure (or almost pure) albumin should also be added (16). From day 3 to day 5 (blastocyst stage), the glucose concentration is increased, as well as the concentration of 20 amino acids. Pyruvate concentration in particular is lower, and protein requirements maintained (16). Change of media at day 3 is appropriate, since the embryo undergoes a series of changes at this time, such as compaction and activation of the embryonic genome (17). As a consequence, sequential media were developed. By contrast, the alternative use of single-step media is based on letting the developing embryos choose the nutrients they need. This way it is possible to minimise the stress caused by different culture environments (13,18). Both types of media are currently used by IVF clinics, even though sequential media seems to be the preferred option (18). DIFFERENCES BETWEEN CURRENT MEDIA AND TRENDS -Differences between sequential and single-step media Previously, two different kinds of media have been introduced: sequential (or two-step) and single-step (or one-step) media. In order to use these media to culture embryos from zygote to blastocyst stage, there are three different protocols available (19): [1] Sequential media protocol. Two media of different compositions are used sequentially. Medium is changed on day 3 of embryo culture. [2] Single-step protocol. Uninterrupted culture using one medium throughout the 5 days. [3] Single-step protocol with renewal. Interrupted culture using the same type of medium throughout the 5 days, including renewal on day 3. Originally, all human embryos were cultured just until day 2-3 before transfer. However, extended culture from zygote to blastocyst has attracted more attention since 1997 (18). The studies by Gardner and Lane (reviewed in 19) or Pool (16) have been the main supporters of the use of sequential media protocols. There are four main arguments in favour of the use of two-step media culture (19): [1] - The energy source required by the preimplantation embryo changes throughout development, from pyruvate and lactate to glucose, but glucose has inhibitory effects on early cleavage stages. [2] - Ethylenediaminetetraacetic acid (EDTA) helps to bypass the two-cell block. Later on, it has an inhibitory effect on blastocyst development and on the inner cell mass (ICM). [3] - Although evidence supports embryo development up to blastocyst stage with no amino acids (AA) present in the media, non-essential AAs (NEAAs), and specially glutamine (Gln) favour embryo growth during cleavage stage. Later on, during blastocyst stage, all 20 AAs stimulate the development of the inner cell mass (ICM). Furthermore, NEAAs also have an stimulation effect on the trophectoderm and the hatching process of the zona pellucida. However, an inhibitory effect on blastocyst development and viability has been observed in the presence of essential AAs (EAAs) during cleavage stage (20). [4] - The chemical breakdown of L-glutamine (Gln) in aqueous solution is the main contributor to the accumulation of ammonia during culture, which can compromise embryo development. Nonetheless, Gln is also beneficial to overcome the two-cell block. The four points mentioned above are easily addressed by using two-step media (21). However, supporters of the one-step medium protocol, in turn, argue that these problems can also be addressed through the use of one single type of medium. First, the inhibitory effects of glucose on early cleavage stages is not absolute. Furthermore, glucose is present in the natural environment of the Fallopian tubes, and there is evidence of culture media with glucose that do not inhibit embryo development (2). The reason behind this is that the inhibitory effect of glucose is determined by the interaction with other substances present in the environment. These interactions can be determined experimentally and adjustments to the concentration of medium components can be made accordingly (19). Furthermore, studies supporting the negative effects of EDTA during late developmental stages of the embryo refer to concentrations of 0.1 mmol/L; the optimal concentration needed to bypass the two-cell block is only between 0.005-0.01 mmol/L, which is insufficient to have any deleterious effect (20). Some authors like Lane and Gardner have suggested leaving out EAAs from the media during early developmental phases (19). By contrast, and after studying the AAs net depletion during human preimplantation embryo development, others such as Leese argue that the most prudent choice would be the use of a mixture with all 20 AAs. This way, the embryo would be able to choose which ones to use by itself (20). The problem of accumulated ammonium from decomposing Gln is solved by using more stable dipeptides of Gln, such as glycol-L-glutamine (GlyGln) or L-alanyl-L-glutamine (AlaGln) (19). Despite the explanation for both types of media addressing the embryo needs throughout early development, there is still a remaining question: which culture medium is more efficient? Even though in recent years multiple studies have dealt with this question, the answer is less than clear. In order to compare one-step protocols with sequential protocols, some studies compare ongoing pregnancy rates, clinical pregnancies or miscarriage rates (22). Other studies also include blastocyst formation per randomized oocyte/zygote (23), or the number and size of blastomeres in certain days, along with the final quality of the embryos (21). But the results of all of them agree in that there is no clear evidence to support either culture medium being better than the other. No significant difference was found between sequential and single medium for ongoing pregnancy rates, clinical pregnancies or miscarriage rates. Regarding embryo development, embryos cultured in one-step medium were found to present significantly more blastomeres (although unequally sized) and lower fragmentation rate in day 2 than those cultured in sequential media. However, no significant difference was observed in the percentage of good quality blastocysts between both groups (21). -Trends in the use of culture media Ever since the beginning, sequential media have been the most popular option. However, the popularity of single-step media has slowly risen within the last decade. This fact is evidenced by the increased offer of commercial media; whereas in 2008 there was only one commercial single-step medium and six sequential media (19), a few years later the number of single-step media available in the market had increased up to three times (20). Not only does the non-renewal single-step media require a reduced level of embryo handling, but it also reduces the chances of damaging or stressing the embryo. This is particularly evident in time-lapse systems, whose medium-related costs are lower. On the other hand, both the one-step medium with renewal approach and sequential protocols avoid excessive accumulation of potentially harmful waste products in the medium. Without reliable evidence supporting one type of medium over the other, it is up to the clinics to decide which one is more suitable. Therefore, further research with larger samples will be needed on comparing the efficiency of embryo culture media. CULTURE MEDIA AND PERINATAL OUTCOMES -Perinatal outcomes and assisted human reproduction Extensive animal research have provide evidence of the link between early embryo development environment and adult diseases. In the same way, in vitro culture may induce epigenetic changes in the embryo with long-term consequences, even if they are not obvious at birth or during early childhood (2). Consistently with these studies, it is known that newborns from assisted reproductive technologies (ART) often have poorer perinatal outcomes (see Table 2) compared to naturally conceived newborns. Usually, ART outcomes have been related to parental underlying medical conditions, preimplantation genetics diagnosis (if performed), cryopreservation and thawing processes, differences in hormonal treatments, laboratory conditions during embryo culture, culture media or the combination of all of them (24). 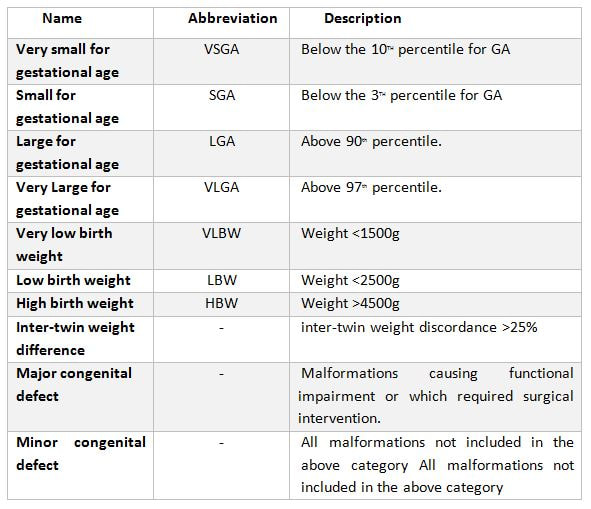 Table 2. Perinatal outcome definitions (26). Up to this day, clinical practice has proven that the use of in vitro culture for human embryos does not imply any major adverse consequences on the offspring. Nevertheless, the population born through ART is still relatively young, being Louise Brown, the first IVF child, only 49 years old. Therefore, the possible adverse repercussions on late childhood or adulthood are still a subject of study (2). -Perinatal outcomes and culture media Within last decade, multiple articles have been published that reveal actual associations between culture media and perinatal outcomes (25,26). Nevertheless, there is certain controversy when choosing between the type (single-step or sequential media) or the brand of the compared media, as well as with sample sizes. Several studies have shown no significant differences between children born after IVF and children conceived spontaneously or by intrauterine insemination (IUI) (24,25). However, there are available data that do show significant disparities in preterm birth rates between groups cultured with different media (25,26,28). Additionally, VitroLife medium seems to present a trend towards newborns large for gestational age (LGA) (25). This medium has been related to significantly higher pregnancy, clinical pregnancy and implantation rates. Simultaneously, Cook medium has been associated with lower birthweight means, higher LBW incidence and higher proportion of single embryo transfers (26). In other cases, when testing VitroLife against other media (SAGE), results indicated no significant differences for any rate; size for gestational age, LBW or even birthweight means between fresh and thawed embryos of both groups showed similar values (29). Lastly, when single-step media SSM and Global were compared, the former exhibited poorer performance than the latter, resulting in lower pregnancy and implantation rates (28). It is clear from previous studies that some culture media underperform in comparison to other media of the same type. As above-mentioned, such lower performance affects important factors like birthweight means. LGA for instance, may cause problems during labour, and LBW has been associated with higher probability of abnormal growth, neurodevelopmental problems and increased incidence of diseases such as obesity or type-2 diabetes (30). Preterm birth, in turn, may lead to the need of incubators and may cause learning disabilities or visual/hearing problems. The most likely reason behind the differences between media efficacy is the disparity in their chemical composition. For instance, whereas VitroLife uses a stable dipeptide of Gln, Cook contains L-Gln, which may be the cause for the higher percentages of LBW observed (31). APPLICATIONS AND NEW APPROACHES IN EMBRYO CULTURE - Applications Approximately, two out of three IVF cycles fail to result in pregnancy, causing significant physical, emotional and financial distress for women undergoing infertility treatments (32). In order to prevent failures, centres have chosen to perform transfer of multiple embryos, which implies a high risk of multiple pregnancy. This strategy increases the chance of developing maternal and infant morbidity; infants from multiple pregnancies are more likely to present low birthweight, and the probability of mothers suffering from miscarriage is also higher. (33). In this regard, the most common maternal complications associated with multiple pregnancies include high blood pressure, preeclampsia, increased likelihood of caesarean section, venous thromboembolism, postpartum hemorrhage and gestational diabetes (34). Considering these data, one major objective in reproductive medicine is trying to find easy, useful and clinically applicable methods to identify embryos with higher probability of implantation. Classic embryo morphology assessment is still the most established method to select embryos for transfer. However, this approach is subjected to the embryologist´s own criteria, and even though there are several scoring systems, it is not a method capable of giving reliable results (35). The recent time-lapse technology provides kinetic information of the embryos and allows the embryologist to have additional criteria for selection. But even with this improvement, more information is needed in order to find the embryo with the highest implantation potential. Nowadays, the OMICS field tries to identify biomarkers for therapeutic and diagnostic development (36). Approaches in embryo assessment based on the analysis of culture media may consider proteomics and metabolomics. Proteomics involves the study of the proteoma, including proteins secreted by embryos in the media. By studying the protein profiles, researchers are able to find altered expression of specific proteins related to important embryo events, like blastocyst development or implantation (37). The use of metabolomics, in turn, allows for the measurement of changes in the level of metabolites present in culture media that are associated with carbohydrate metabolism and amino acid turnover. The concentration of these metabolites can be used as main indicators for embryo potential. Metabolites such as glucose or lactate can effectively be associated with embryo development. Because metabolism is the final product of gene expression, the study of metabolism may be useful compared to the other OMICS, and cell function may be more accurately reflected (37). Additionally, not only metabolic flow is regulated by genetic expression, environmental stress and metabolites can be measured more precisely (39). Future directions of OMIC research should not only be focused on trying to elucidate which embryo presents the most suitable profile for implantation, but also on trying to reduce the cost of required technology and turn it affordable for daily clinical practice. - New approaches for embryo culture Culture media composition has been modified in the past recent years in order to achieve better physiological conditions. By contrast, research on embryo biophysical requirements has not developed equally fast (40). When developed in vivo, not only embryos are exposed to changing fluid chemical composition, but to mechanical stimulation (41,42,43). In addition, changes in the type of culture platform may lead to modifications in the environment immediately surrounding the embryo through regulation of chemical gradients. Thus, research on different new technologies may assist in improving embryo development (44,45). Types of culture systems
Microdrop systems have the benefit of the growth factors, with a typical drop setting of 10-50 μL. Ultramicrodrop is a variant that works with volumes of 1.5-2.0 μL, but it still presents severals typical issues related to working with small volumes (evaporation, osmolality, potential toxicity, embryo loss...) (55). A different model is conformed by the use of volumes of sub-μL (nL) of media and a vertical channel-containing polydimethylsiloxane (PDMS) culture chip (56). This allows the embryo to benefit from both reduced media volume and space. On the other hand, these same features makes the embryo difficult to recover. The Microwells system creates a microenvironment that offers a potentially increasing surface area and simultaneously a reduced space between embryos. The most popular microwell, called “well of the well” (WOW), was developed by Vajta (2000), and it has since been validated by several studies in several species (57). Initially, microwells needed to be made manually, but there currently exist available WOW systems made of polystyrene. Microchannels are related to the idea of increasing the surface area adjacent to the embryo, rather than just a single point of contact within a Petri dish. A different approach of microchannels is using glass capillary tubes filled with media (58).
Vibration systems have been developed due to the estimation that, in vivo, embryos are exposed to vibrations of 5-20 Hz in response to the ciliated epithelium of the oviduct (62). Data from different studies have shown that even short periods of gentle vibrations during oocyte maturation or early fertilization events may be of benefit to embryo quality (63,64). Finally, controlled fluid systems have been developed based on the premise that embryos, just like other cell types, can detect the shear stress (an endothelial concept) of the fluid flow (65). Excessive stress could damage the blastomeres, affect signaling pathways and cause embryo degeneration (66). CONCLUSIONS Culture media have experienced a significant development in ART, from primordial cultures like simple salt solutions supplemented with glucose and egg yolk, including changes in embryo energy sources, until more complex solutions closer to in vivo reproductive fluids. This evolution of culture media has been possible thanks to research on embryo metabolism. This allowed to find out the nutritional requirements of the embryo for every stage, thereby making it possible to develop sequential media. The development of the alternate philosophy (“let the embryo choose”) has in turn offered other advantages, such as the reduction of stress caused by different microenvironments. Both approaches have defenders and detractors, all of them with arguments to support one over the other. However, it is not yet clear which media offers the best “in vitro environment” for the embryos. Single media have gained supporters partly due to their advantages regarding reduced manipulation. This is related to time-lapse technology, even though the classical sequential media or the “renew single media” option are widely employed, too. The usage of these media is justified by the potentially deleterious effects of the accumulation of harmful waste substances. On the other hand, different ART-related procedures, including embryo culture, seem to also affect the offspring, showing poorer perinatal outcomes in comparison to naturally conceived newborns. It remains unclear whether this is derived from the culture media used or from combinations of other parameters. However, current data indicate that media do not seem to entail any major adverse consequences on the offspring. Even so, caution should be exercised. Different chemical proportions and the quality of molecular composition could explain the differences observed in some parameters like LBW between distinct commercial brands. It is important to have more information about the role of culture media in adverse perinatal outcomes. For this purpose, not just the chemical composition but also the concentrations of commercialised media should be disclosed to facilitate research on this subject. It seems obvious that modifications in the platforms used for culture media may influence embryo development. Dynamic systems are a new field of research, which may potentially help understand the physiological requirements of the embryo. However, it might result difficult to eventually improve these strategies due to extra expenses. In order to achieve so, such new devices need to count on the companies´ trust, since they are the actual responsibles for making it possible to spread technology throughout the clinical community. REFERENCES 1- Nowotschin S, Ferrer-Vaquer A, Hadjantonakis AK. Imaging Mouse Development with Confocal Time-Lapse Microscopy. Methods Ezymol. 2010;476:351-377. 2- Chronopoulou E, and Harper JC. IVF culture media: past, present and future. Hum Reprod Update. 2015;21(1):39-55. 3- Yin TL, Zhang Y, Li SJ, et al. Culture media influenced laboratory outcomes but not neonatal birth weight in assisted reproductive technology. J Huazhong Univ Sci Technolog Med Sci. 2015;35(6):932-937. 4- Sirard MA. The influence of in vitro fertilization and embryo culture on the embryo epigenetic constituents and the possible consequences in the bovine model. J Dev Orig Health Dis. 2017;8(4):411-417. 5- Lewis WH and Gregory PW. Cinematographs of living developing rabbit-eggs. Science. 1929;69:226–229. 6- Kuhl W. Untersuchungen uber die cytodynamik der furchung und fruhentwicklung des eis der weissen maus. Abb Senchenb Naturforsch Ges. 1941;456:1–17. 7- Hammond J. Recovery and culture of tubal mouse ova. Nature. 1949;163:28–28. 8- Whitten WK. Culture of tubal mouse ova. Nature. 1956;177:96–96. 9- Brinster, R.L. Studies on the development of mouse embryos in vitro. II. The effect of energy source. J. Exp. Zool. 1965;158:59–68. 10- Edwards RG, Bavister BD, Steptoe PC. Early stages of fertilization in vitro of human oocytes matured in vitro. Nature. 1969;221:632–635. 11- Steptoe PC, Edwards RG, and Purdy JM. Human blastocysts grown in culture. Nature. 1971;229:132–133. 12- Steptoe PC, and Edwards RG. Birth after the reimplantation of a human embryo. Lancet. 1978;2:366–366. 13- Yao T, Asayama Y. Human preimplantation embryo culture media: past, present, and future. J. Mamm. Ova Res. 2016;33(1):17–34. 14- Lawitts JA, and Biggers JD. Culture of preimplantation embryos. Methods Enzymol. 1993;225:153–164. 15- Ho Y, Wigglesworth K, Eppig JJ and Schultz R.M. Preimplantation development of mouse embryos in KSOM: augmentation by amino acids and analysis of gene expression. Mol. Reprod. Dev. 1995;41:232–238. 16- Pool TB. An update on embryo culture for human assisted reproductive technology: media, performance, and safety. Semin Reprod Med. 2005;23(4):309-318. 17- Quinn P. Culture systems: sequential. Methods Mol Biol. 2012;912:211-30. 18- Vajta G, Rienzi L, Cobo A, and Yovich J. Embryo culture: can we perform better than nature? Reprod Biomed Online. 2010;20(4):453-469. 19- Biggers, JD and Summers MC. Choosing a culture medium: making informed choices. Fertility and Sterility 2008, Volume 90, Issue 3, 473 – 483. 20- Gruber, I, and Matthias K, Embryo Culture Media for Human IVF: Which Possibilities Exist? Journal of the Turkish German Gynecological Association 12.2 (2011): 110–117. 21- Paternot G, Debrock S, D´Hooghe TM, and Spiessens C. Early Embryo Development in a Sequential versus Single Medium: A Randomized Study. Reproductive Biology and Endocrinology : RB&E 8 (2010): 83. 22- Dieamant, F, Petersen CG, Mauri AL, et al. Single versus Sequential Culture Medium: Which Is Better at Improving Ongoing Pregnancy Rates?, A Systematic Review and Meta-Analysis.” JBRA Assisted Reproduction 21.3, 2017: 240–246. 23- Sfontouris, IA, Martins WP, & Nastri CO, et al. Blastocyst Culture Using Single versus Sequential Media in Clinical IVF: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Journal of Assisted Reproduction and Genetics 33.10 (2016): 1261–1272. 24- Marianowski P., Dabrowski FA, Zygula A, et al. Do We Pay Enough Attention to Culture Conditions in Context of Perinatal Outcome after In Vitro Fertilization? Up-to-Date Literature Review. BioMed Research International, vol. 2016, Article ID 3285179, 6 pages, 2016. doi:10.1155/2016/3285179. 25- Dumoulin JC, Land JA, Van Montfoort AP, et al. Effect of in vitro culture of human embryos on birthweight of newborns Human Reproduction, Volume 25, Issue 3, 1 March 2010, Pages 605–612. 26- Nelissen EC, Van Montfoort AP, Coonen E, et al. Further evidence that culture media affect perinatal outcome: findings after transfer of fresh and cryopreserved embryos. Human Reproduction, Vol.27, No.7 pp. 1966–1976, 2012 doi:10.1093/humrep/des145. 27- Sandi-Monroy NL, Winkle T, Huzurudin N, et al. Does in vitro culture of human preimplantation embryos negatively affect perinatal outcomes? American Society for Reproductive Medicine Vol. 102, No. 3, Supplement, September 2014 pages 306-307. 28- Bouillon C, Léandri R, Deschet L, et al. Does Embryo Culture Medium Influence the Health and Development of Children Born after In Vitro Fertilization?. PLoS ONE 11.3 (2016): e0150857. 29- Fang Gu, Mingfen Deng, Jun Gao et al. The effects of embryo culture media on the birthweight of singletons via fresh or frozen-thawed embryo transfer: a large scale retrospective study. BMC Pregnancy and Childbirth, 2016, 16:270 DOI 10.1186/s12884-016-1077-7. 30- World Health Organization (WHO). Preterm birth. Fact sheet, November 2017 31- Zandstra H, Van Montfoort APA,and Dumoulin JCM. Does the type of culture medium used influence birthweight of children born after IVF?, Human Reproduction, Volume 30, Issue 3, 1 March 2015, Pages 530–542, https://doi.org/10.1093/humrep/deu346. 32- SART. Assisted reproductive technology success rates. National summary and fertility clinic reports. Centers for disease control, USA, 2006. 33- Lieberman B, Ali R, and Rangarajan S. Towards the elective replacement of a single embryo transfer (eSET) in the United Kingdom. Human Fertility, vol.10, no 2pp.123-127, 2007. 34- Walker MC, Murphy KE, Pan S, et al. Adverse maternal outcomes in multifetal pregnancies. American Journal of Obstetrics and Gynecology, vol.111, no.11, pp.1294-1296, 2004. 35- Rocha JC, Felipe Passalia, Felipe D. Matos, Marc P. Maserati Jr, Bruna L. Cardoso, Andrea C. Basso and Marcelo F.G. Nogueira. Methods for assessing the quality of mammalian embryos. How far we are from the gold standard?. JBRA. Assist Reprod. 2016 Jul-Sep; 20(3): 150-158. 36- Zhang-Zhi Hu, Hongzham Huang, Cathy H. Wu, Mira Jung, Anatoly Dritschilo, Anna T.Riegel and Anton Wellstein. Omics-Based on Molecular Target and Biomarker identification. Methods Mol Biol. 2011;719:547-571. 37- Domínguez F, Gadea B, Esteban FJ, Horcajadas JA, Pellicer A, Simón C. Comparative protein profile analysis of implanted vs non-implanted human blastocysts. Human Reprod. 2008; 23: 1993-2000. 38- Urbanczyk-Wochniak E, Luedemann A, Kopka J, Selbig J, Roessner- Tunali U, Willmitzer L, Fernie AR. Parallel analysis of transcript and metabolic profile. EMBO Rep 2003; 4:989-992. 39- Johnson HE, Broadhurst D, Goodacre R, Smith AR. Metabolic fingerprinting of salt tomatoes. Phytochemestry 2003; 62: 919-28. 40- Gary D. Smith, Suichi Takayama, Jason E. Swain.Rethinking in Vitro Embryo Culture: New developments in Culture Platforms and Potential to Improve Assisted Reproductive Technologies. Biology of Reproduction, Volume 86, Issue 3, 1 March 2012,62,1-10. 41- Westrom L, Mardh PA, Mecklenburg CV, Hakansson CH. Studies on ciliated epithelia of the human genital tract. II. The mucociliary wave pattern of fallopian tube epithelium. Fertil Steril 1977;28: 955 961. 42- Paltieli Y, Weichselbaum A, Hoffman N, Eibschitz I, Kam Z. Laser scattering instrument for real time in-vivo measurement of ciliary activity in human fallopian tubes. Hum Reprod 1995; 10: 1638 1641. 43- Fauci L, Dillon R. Biofluidmechanics of reproduction. Annu Rev Fluid Mech 2006;38: 371 394. 44- Swain JE, Smith GD. Advances in embryo culture platforms: novel approaches to improve preimplantation embryo development through modifications of the microenvironment. Human Reprod Update 2011; 17: 541557. 45- Smith GD, Swain JE, Bormann CL. Microfluidics for gametes, embryos, and embryonic stem cells. Semin Reprod Med 2011;29:5 14. 46- Reed ML, Woodward BJ, Swain JE. Single or group culture of mammalian embryos: the verdict of the literature. J Reprod Stem Cell Biol 2011;2:77 87. 47- Borman C, Swain J, Ni Q, Kennedy R, Smith G. Preimplantation embryo secretome identification. Fertil Steril 2006; 86:s116. 48- Katz-Jaffe MG, Schoolcraft WB, Gardner DK. Analysis of protein expression (secretome) by human and mouse preimplantation embryos. Fertil Steril 2006; 86:678 685. 49- Cortezzi SS, Garcia JS, Ferreira CR, Braga DP, Figueira RC, Jaconelli A, Jr, Souza GH, Borges E, Jr, Erbelin MN. Secretome of the preimplantation human embryo by bottom- up label- free proteomics. Anal Bioanal Chem 2011; 401:1331 1339. 50- Richter KS. The importance of growth factors for implantation embryo development and in- vitro culture. Curr Opin Obstet Gynecol 2008; 20: 292 304. 51- Lane M, Garner DK. Effect of incubation volume and embryo density on the development and viability of mouse embryos in vitro. Human Reprod 1992; 7: 558 562. 52- Donnay I, Van Langendonckt A, Auquier P, Grisart B, Vansteenbrugge A, Massip A, Dessy F. Effects of co-culture and embryo number on the in vitro development of bovine embryos. Theriogenology 1997; 47: 1549 1561. 53- Spindler RE, Crichton EG, Agca Y, Loskutoff N, Critser J, Gardner DK, Wildt DE. Improved felid embryo development by group culture is maintained with heterospecific companions. Theriogenology 2006; 66: 82 92. 54- Moessner J, Dodson WC. The quality of human embryo growth is improved when embryos are cultured in groups rather than separately. Fertil Steril 1995; 64: 1034 1035. 55- Ali J. Continuos ultra micro drop culture yields higher pregnancy and implantation rates than either large- drop culture or fresh- medium replacement. Clin Embryol (Online) 2004; 7: 1 23. 56- Melin J, Lee A, Foygel K, Leong DE, Quake SR, Yao MW. In vitro embryo culture in defined, sub-microliter volumes. Dev Dyn 2009; 238:950 955. 57- Vaita G, Peura TT, Holm P, Paldi A, Greve T, Trounson AO, Callesen H. New method for culture of zona -included or zona-free embryos: the well of the well (WOW) system. Mol Reprod Dev 2000; 55: 256 264. 58- Thouas GA, Jones GM, Trounson AO. The “GO” system - a novel method of micro culture for in vitro development of mouse zygotes to the blastocyst stage. Reproduction 2003; 126: 161 169. 59- Trimarchi JR, Liu L, Smith PJ, Keefe DL. Non invasive measurements of potassium efflux as an early indicator of cell death in mouse embryos. Biol Reprod 2000; 63:851-857. 60- Lane M, Hooper K, Gardner DK. Effect of essential amino acids on mouse embryo viability and ammonium production. J Assist Reprod Genet 2001; 18: 519 525. 61- Matsumura K, Hayashi N, Kuroda Y, Takiue C, Hirata R, Takenami M, Aoi Y, Yohshioka N, Habara T, Mukaida T, Naruse K. Improved development of mouse and human embryos using a tilting embryo culture system. Reprod Biomed Online 2010; 20: 358 364. 62- Palieli Y, Weichselbaum A, Hoffman N, Eibschitz I, Kam Z. Laser scattering instrument for real time in -vivo measurement of ciliary activity in human fallopian tubes. Hum Reprod 1995; 10: 1638 1641. 63- Mizobe Y, Yoshida M, Miyoshi K. Enhancement of cytoplasmic maturation of in vitro-matured pig oocytes by mechanical vibration.J Reprod Dev 2010; 56: 285 290. 64- Isachenko E, Maettner R, Isachenko V, Roth S, Kreienberg R, Sterzik K. Mechanical agitation during the in vitro culture of human pre-implantation embryos drastically increases the pregnancy rate. Clin Lab 2010; 56: 569 576. 65- Xie Y, Wang F, Zhong W, Puscheck E, Shen H, Rappolee DA. Shear stress induces preimplantation embryo death that is delayed by the zona pellucida and associated with stress-activated protein kinase-mediated apoptosis. Biol Reprod 2006; 75: 45 55. 66- Xie Y, Wang F, Puscheck EE, Rappolee DA. Pipetting causes shear stress and elevation of phosphorylated stress-activated protein kinase/jun kinase in preimplantation embryos. Mol Reprod Dev 2007; 74: 1287 1294. |
Entries
March 2019
Categories
All
 2016-2019. All Rights Reserved by Embryologist Media. This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License . |
Embryologist Media
