|
Authors: María Caballero Sastre & Raquel Pillado González “Currently, performing any cryopreservation technique results in some degree of damage to the sperm.“ 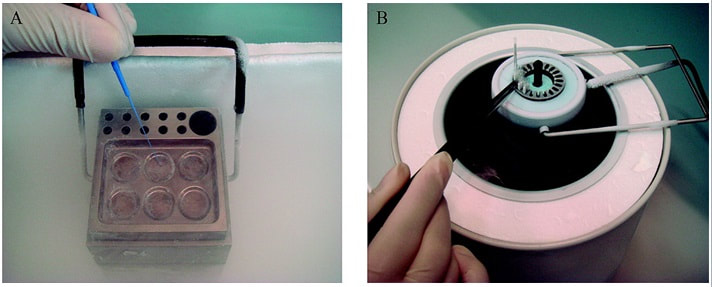 Figure 1. Solid Surface Vitrification (A) and slow cooling equipment (B) (1). THE HISTORY OF SPERM CRYOPRESERVATION Human semen cryopreservation has a long history that begins at the end of the nineteenth century. After prior observations on sperm surviving cooling at very low temperatures (-150º C) (2), Mantegazza (1866) first suggested the idea of human sperm banks (3). Years later, Mantegazza proved it was possible to extend human sperm lifespan up to four days by cooling at moderate sub-zero temperatures (-17º C) (4). Although a few extra days of storability does not make much difference in practical terms, this was the starting point of further research aimed to develop techniques that would allow for longer storage periods. The next leap forward in this field was the discovery of the cryoprotectant properties of glycerol in 1949. This molecule proved to be an effective cryoprotectant agent (CPA) when combined with bovine sperm, and allowed for the development of new cryopreservation methods through which sperm could preserve their motility and fertilization capabilities even after the freezing-thawing process (5). However, during the following years this new technique was primarily applied to cryopreservation of farm animal semen rather than human semen (6). In 1953, at the University of Iowa, the first case of live birth from cryopreserved sperm was reported (7). This successful birth, along with reports of high survival percentage (67%) of human sperm after cryopreservation, popularized the utilization of glycerol with human semen. The most common method to use glycerol was the protocol described by Bunge and colleagues, which stated the processing of sperm in a 10%-glycerol solution before freezing it with dry ice (7). A decade later, the use of liquid nitrogen was introduced for long-term sperm cryopreservation. This new method led to the progressive normalization of sperm freezing as a widespread practice in healthcare (8). With the availability of long-term storage and the extended use of sperm freezing, new methods and variants were developed over time, such as slow freezing, fast freezing, LN2 vapours or lyophilization, which will be later described. Nowadays, cryopreservation is routinely used in most assisted reproduction centres for numerous reasons:
It is important to possess a clear understanding of the semen cryopreservation process due to its current importance in clinical and research environments. Modern assisted reproduction practices are unthinkable without this tool. CRYOPRESERVATION TECHNIQUES Before being frozen, a sperm sample needs to be appropriately processed in order to separate sperm cells from the seminal plasma. This helps increase the concentration of high quality spermatozoa for a later use. Different techniques for sperm selection have been reviewed in our previous post. Cryoprotectants The process of cryopreservation may involve irreversible cellular damage due to a change in the osmotic balance (11). Upon freezing of the extracellular water, the solute concentration increases in this fraction. As a result, the intracellular water is transported out of the cell to restore the osmotic balance, which may lead to cell dehydration and irreversible membrane damage (12, 13). Consequently, CPAs need to be incorporated along the sperm sample following processing. These molecules will protect spermatozoa by reducing intracellular ice formation and by decreasing the freezing point of the medium and the solute concentration present in the extracellular environment (14). There are two kinds of cryoprotective agents: permeable and non-permeable. The permeable ones, such as glycerol, dimethyl sulfoxide or ethylene glycol, protect the intracellular structures and biomolecules. Non-permeable agents, like sucrose, polysaccharides and some proteins, contribute to keeping the osmotic equilibrium, thus preventing cellular dehydration (11). Procedures - Cryopreservation in liquid nitrogen (LN2) This is the most commonly used method to cryopreserve sperm. It can be subdivided into three methods:
- Cryopreservation in microdroplets Microdroplets are sometimes used in the clinic to cryopreserve sperm in small volumes. This is preferred in cases such as epididymal sperm aspiration. Around 50-100 μL of the sperm-CPA solution are placed on a dry ice plate up to freezing (Fig. 2); droplets formed in such a way are then kept in vials and plunged into LN2 (18).  Figure 2. Cryopreservation in droplets. Dry ice plate used to cool down samples that form sperm droplets (in yellow). - Vitrification This technique consists of cooling the sample at ultrarapid rates, so that the water solidifies (vitrifies) as a glass-like structure rather than forming ice crystals (19). However, even though vitrification should cause minimal damage, this is not always the case in clinical practice. One of the most frequent problems is the requirement for very high CPA concentrations that sperm do not tolerate well (20). Despite this, some studies have demonstrated it is possible to perform vitrification without using CPAs (20-22). Also, the large volume of sperm typically used impedes the cooling of the sample at the appropriate speed, causing ice formation (4). Despite these limitations, a vast proportion of clinics use vitrification as a routine practice due to its practical advantages. In recent years, a new variant of this technique has been developed. This evolved version is called Solid Surface Vitrification (SSV) (23, 24). For this procedure, the sample or tissue is directly exposed to a metal surface previously precooled at -160º C before the use of LN2 (Fig. 1). This method prevents the apparition of nitrogen bubbles and evaporation that would slow the cooling rate (24). This technique has been previously applied to animal mature oocytes and human gametes and embryos, yielding successful results (23, 24). OUTCOME COMPARATIVE BETWEEN TECHNIQUES Because every cryopreservation technique shows different advantages and disadvantages, it is important to bear in mind how sperm will be affected during the thawing process. Parameters such as motility, viability, morphology and DNA integrity are evaluated accordingly. Regarding LN2 techniques, no studies so far have directly compared post-thaw sperm quality following slow and rapid freezing. The literature shows agreement between studies on the main problem in both processes, which is controlling cooling rates (reviewed in 16). If the cooling rate is too fast, ice crystals may be formed inside the cells. By contrast, if it is too slow, the result could be cell contraction due to osmotic stress (25). This issue can be faced by using an automated programmable freezer, but only when keeping a large number of samples (26). Even though several reports have compared fast and slow freezing in animal reproduction, conclusions are controversial. Some studies in semen from horse (27) and buffalo (28) have reported better results when using fast thawing, whereas other authors have found no difference between fast and slow thawing rates (29). Nevertheless, it seems clear that the critical point lays on thawing matching the freezing process. Considering rapid freezing, thawing is recommended to be also carried out at a fast rate to avoid formation of intracellular ice crystals. Likewise, for the slow-rate cooling procedure, the sample needs a slow thawing protocol, since cells need more time to rehydrate (30). On the other hand, all studies on the use of LN2 show similar or slightly better results regarding the aforementioned parameters when using nitrogen vapours (31-33). These results, however, are obtained after short-term storage of samples in nitrogen vapours of up to three months. When stored for longer, sperm quality decreases. Consequently, this method is only recommended for short-term storage (further research would be needed in order to support its application for long-term storage) (34). Upon comparison between vitrification and LN2 techniques, different results can be highlighted. Certain authors determined that results of sperm parameters such as motility, viability and normal morphology were similar between vitrification and rapid freezing techniques (21). On the contrary, different results were found for DNA fragmentation rates. Whilst some groups found that DNA fragmentation was significantly higher for the rapid freezing technique (12, 21) or for LN2 vapours (24), other groups obtained contradictory or uncertain results (35, 36). For instance, DNA fragmentation has been observed to increase over time when analyzing semen 6h after thawing, compared to recently-thawed samples (35). Despite the different results obtained, vitrification shows important advantages compared to other available techniques. Some of these advantages are: 1) unnecessary use of CPAs; 2) the technique is simpler and faster compared to conventional slow freezing, due to the fact that once the sample is kept in a proper container (such as cryoloop or straws), it is rapidly plunging into LN2 to be stored; 3) no requirement for programmable freezers; 4) the sample is free of seminal plasma and potential pathogens (vitrification is usually performed after swim-up); 5) no requirement for post-thaw processing (reviewed in 6). TRENDS IN IMPROVING SEMEN CRYOPRESERVATION Currently, performing any cryopreservation technique results in some degree of damage to the sperm (4). The severity may differ depending on the initial quality of the sample, being greater in poor quality semen. Luckily, the application of ICSI allows for the successful use of low quality sperm (if necessary) even after having been cryopreserved. Side effects of cryopreservation on sperm include reduced motility, vitality, viability and increased DNA damage. Although motility is the most affected parameter, DNA damage entails greater detrimental effects regarding embryo viability (6). The majority of the harm produced by cryopreservation occurs during the freezing and thawing phases, the crucial moments being between -15º C and -60º C. It is worth mentioning that a considerable damage is produced by CPAs themselves; these agents cause oxidative stress that derives in the formation of reactive oxygen species (ROS) (8, 10, 37). Furthermore, these components affect the polyunsaturated fatty acids in plasmatic membranes due to lipid peroxidation (10). Consequently, phospholipids reservoirs such as egg yolk are usually added to the freezing media. As a more direct countermeasure, several current research lines look into numerous antioxidant components in order to be added to freezing media, thus avoiding the damage caused by ROS. Examples of these antioxidants are TAT-peroxiredoxin-2 fusion protein, quercetin or melatonin. Preliminary research indicates that their presence results in higher motility and viability rates post-thawing, along with a reduction of intracellular ROS levels (8, 10). Other approaches consider the utilization of protocols that may directly disregard the use of CPAs, such as certain vitrification protocols previously mentioned. Sperm freezing entails other associated problems, too, such as the loss of chromatin and acrosome integrity that had been observed post-thaw. Recent data have reported differences in the levels of DNA and acrosome integrity after cryopreservation depending on the freezing technique used (24). The difference in DNA integrity levels is suspected to be due to the cold shock faced by the samples (24). This issue could be amended by the development of media able to preserve sperm without freezing, an avenue that is currently being pursued. For instance, Riel and colleagues have reported that the use of an electrolyte-free medium for short-time (1 week) storage of semen yields better levels of DNA integrity in comparison to traditional cryopreservation. If the storage period capacity could be further improved, this might become a rather attractive alternative (38). Lyophilization or freeze drying is an experimental technique that has been proven less harmful to the DNA (4, 10, 38). In order to perform this method, the sample must be cooled below the triple point of water (Fig. 3). At this temperature solid water (ice) sublimates when the pressure is decreased and exits the cell, leaving it fully dehydrated (4, 10). However, this process irreversibly damages the sperm membrane, thus resulting in non-motile or even non-viable (dead) sperm. Nevertheless, studies on mouse sperm have shown that lyophilized spermatozoa can be used for fertilization with the assistance of ICSI (39). Although the first attempts to use lyophilization on human sperm were in the 50s, today there is still a lacking protocol for this technique that is able to preserve both sperm motility and viability (4). 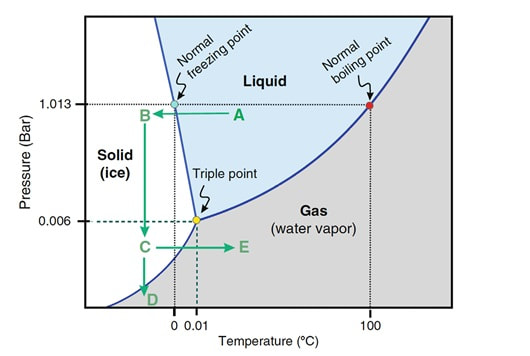 Figure 3. Water phase diagram showing the relation of the conditions of temperature and pressure for freeze-drying (not in scale). Samples are frozen by reducing the temperature (A to B) and then the pressure is also reduced by aspiration (creating vacuum) so the sample lies below the ‘triple’ point (C) for both temperature and pressure (this is the point where all states co-exist). From here on the sample is subjected to a controlled increase of temperature or to a further decrease of pressure to sublimate ice (for detailed current sperm freeze-drying protocols, see (40) and (41)). Modified from (41). The main advantages of freeze drying are: the possibility to preserve spermatozoa with high DNA integrity for at least a year and a half (39), the inactivation of viruses that may be present and the fact that liquid nitrogen is not required. Additionally, samples can be stored at 4º C and transported at room temperature (4). To date, this method still remains experimental regarding humans, due to the lack of actual data on the matter (4, 10). CONCLUSIONS Cryopreservation has gone a long way. Its use in reproductive medicine got to revolutionize the horizon for infertile couples. New doors opened decades ago, and it is fair to reason new ones will open in the near future. Egg donation, social freezing, embryo cryopreservation. Times and timing have changed for patients, and clinics and reproduction centres faced the need for evolution in order to cope with rising approaches. In spite of the variety of options for semen cryopreservation, all of them present their own limitations. Continuous research allows for the discovery of new ways to correct these flaws; however, there lies a long path ahead, and further studies will be required before any improvement can be incorporated to routine practice. REFERENCES
1. Wang X, Catt S, Pangestu M and Temple-Smith P. Live offspring from vitrified blastocysts derived from fresh and cryopreserved ovarian tissue grafts of adult mice. Soc Reprod Fert.2009;138(3): 527–535. 2. Varghese AC, Nandi P, Mahfouz R, Athayde KS, Agarwal A. Human Sperm Cryopreservation. In: Varghese, AC., Nandi, P., Mahfouz, R., Athayde, KS., Agarwal A, editor. ANDROLOGY LABORATORY MANUAL [Internet]. Cleveland Clinic. 2014; p.196–206. 3. Bunge, GR., & Sherman, KJ. Fertilizing Capacity of Frozen Human Spermatozoa. Nature.1953; 172(4382):767-8. 4. Mocé E, Fajardo AJ, Graham JK. Human sperm cryopreservation. EMJ. 2016;1:86–91. 5. Polge C, Smith AV, and Parkes AS. Revival of Spermatozoa after Vitrification and Dehydration at Low Temperatures Nature.1949; 164, 666. 6. Sharma R, Kattoor AJ, Ghulmiyyah J, Agarwal A, Sharma R, Kattoor AJ, et al. Effect of sperm storage and selection techniques on sperm parameters. Syst Biol Reprod Med. 2015;61(1):1–12. 7. Bunge RG, Sherman JK. Fertilizing capacity of frozen human spermatozoa. Nature. 1953;172:767–768. doi: 10.1038/172767b0. 8. Rozati H, Handley T, Jayasena C. Process and Pitfalls of Sperm Cryopreservation. J Clin Med. 2017;6(89):1–13. 9. Tiwari A, Tekcan M, Sati L, Murk W, Stronk J. A new media without animal component for sperm cryopreservation : motility and various attributes affecting paternal contribution of sperm. J Assist Reprod Genet. Journal of Assisted Reproduction and Genetics; 2017;34:647–57. 10. Karimfar MH, Niazvand F, Haghani K, Ghafourian S, Shirazi R, Bakhtiyari S. The protective effects of melatonin against cryopreservation-induced oxidative stress in human sperm. Int J Immunopathol Pharmacol. 2015;28(1):69–76. 11. Sieme H, Oldenhof H, Wolkers WF. Mode of action of cryoprotectants for sperm preservation. Anim Reprod Sci [Internet]. Elsevier B.V.; 2016;1–14. 12. Elliott GD, Wang S, Fuller BJ. Cryobiology Cryoprotectants : A review of the actions and applications of cryoprotective solutes that modulate cell recovery from ultra-low temperatures. Cryobiology [Internet]. Elsevier Ltd; 2017;76:74–91. 13. Royere D, Barthelemy C, Hamamah S, Lansac J. Cryopreservation of spermatozoa: a 1996 review. Hum Reprod Update. 1996;2(6):553-9. 14. Thachil JV, and Jewett, MA. Preservation techniques for human semen. Fertil Steril. 1981;35:546–8. 15. Holt, WV. Basic aspects of frozen storage of semen. Anim Reprod Sci. 2000; 62(1–3):3-22. 16. Sherman, JK. Cryopreservation of human semen. In: Handbook of the Laboratory Diagnosis and Treatment of Infertility. 1990; Keel B. and Webster BW, eds. Boca Raton, Fla, USA: CRC Press, pp. 229–59. 17. Fountain D, Ralston M, Higgins N, Gorlin, J, Uhl L, Wheeler C, et al. Liquid nitrogen freezers: a potential source of microbial contamination of hematopoietic stem cell components. Transfusion, 1997;37:585–91. 18. Abdelhafez F, Mohamed B, El-nashar S, Sabanegh E, Desai N. Techniques for cryopreservation of individual or small numbers of human spermatozoa : a systematic review. Hum Reprod. 2018;15(2):153–64. 19. Kuleshova LL, Lopata A. Vitrification can be more favorable than slow cooling. Fertil Steril. 2002;78(3):449-54. 20. Isachenko E, Isachenko V, Katkov II, Dessole S, Nawroth F. Vitrification of mammalian spermatozoa in the absence of cryoprotectants: from past practical difficulties to present success. Reprod Biomed Online. 2003;6(2):191-200. 21. Agha-Rahimi A, Khalili MA, Nabi A, Ashourzadeh S. Vitrification is not superior to rapid freezing of normozoospermic spermatozoa: effects on sperm parameters, DNA fragmentation and hyaluronan binding. Reprod Biomed Online. 2014;28(3):352-8. 22. Nawroth F, Isachenko V, Dessole S, Rahimi G, Farina M, Vargiu N et al. Vitrification of human spermatozoa without cryoprotectants. Cryo Letters. 2002;23:93–102. 23. Kamath, MS., Muthukumar, K., Appendix B: Solid Surface Vitrification. Methods Mol. Biol.2017;1568:297-307. 24. Rahiminia T, Hosseini A, Anvari M, Ghasemi-esmailabad S, Talebi AR. Modern human sperm freezing: Effect on DNA , chromatin and acrosome integrity. Taiwan J Obs Gynecol. 2017;56(Feb):472–6. 25. Said TM, Gaglani A, Agarwal A. Implication of apoptosis in sperm cryoinjury. Reprod Biomed Online. 2010;21(4):456-62. 26. Pugliesi G, Fürst R, Carvalho GR. Impact of using a fast-freezing technique and different thawing protocols on viability and fertility of frozen equine spermatozoa. Andrologia. 2014;46(9):1055-62. 27. Fürst R, Carvalho GR, Fürst MCO, Ruas JRM, Borges AM, et al. Efeito do resfriamento do sêmen eqüino sobre sua congelabilidade. Arq Bras Vet Zootec. 2005;57:599–607. 28. Shah SA, Andrabi SM, Qureshi IZ. Effect of equilibration times, freezing, and thawing rates on post-thaw quality of buffalo (Bubalus bubalis) bull spermatozoa. Andrology. 2016;4(5):972-6.b. 29. Vidament M, Yvon JM, Couty I, Arnaud G, Nguekam- Feugang J, et al. Advances in cryopreservation in modified INRA 82. Anim Reprod Sci 68:201–218. 30. Mazur P. Basic concepts in freezing cells. In: Proc. 1st International Conf. Deep Freezing Boar Semen. Uppsala, Sweden, 2005;91–111. 31. Amesse LS, Srivastava G, Uddin D, and Pfaff-Amesse T. Comparison of cryopreserved sperm in vaporous and liquid nitrogen. J Reprod Med. 2003;48:319–24. 32. Saritha KR, and Bongso, A. Comparative evaluation of fresh and washed human sperm cryopreserved in vapor and liquid phases of liquid nitrogen. J Androl. 2001;22:857–62. 33. Satirapod C, Treetampinich C, Weerakiet S, Wongkularb A, Rattanasiri S, et al. Comparison of cryopreserved human sperm from solid surface vitrification and standard vapor freezing method: on motility, morphology, vitality and DNA integrity. Andrologia. 2012;44(Suppl. 1):786–790. 34. Lim JJ, Shin TE, Song S, Bak CW, Yoon TK and Lee DR. Effect of liquid nitrogen vapor storage on the motility, viability, morphology, deoxyribonucleic acid integrity, and mitochondrial potential of frozen-thawed human spermatozoa. Fertil Steril. 2010;94:2736–41. 35. Gosalvez J, Nunez R, Fernandez JL, Lopez-Fernandez C, Caballero P. Dynamics of sperm DNA damage in fresh versus frozen–thawed and gradient processed ejaculates in human donors. Andrologia- 2011;43:373–377. 36. Isachenko E, Isachenko V, Katkov II, Rahimi G, Schondorf T, et al. DNA integrity and motility of human spermatozoa after standard slow freezing versus cryoprotectant-free vitrification. Hum. Reprod.2004;19:932–939. 37. Wu Y. Successful delivery derived from cryopreserved rare human spermatozoa with novel cryopiece. Am Soc Androl. 2017;5:832–7. 38. Gianaroli L et al. DNA integrity is maintained after freeze-drying of human spermatozoa. Fertil Steril. 2012;97(5):1067-73. 39. Ward MA, Kaneko T, Kusakabe H, Biggers JD, Whittingham DG and Yanagimachi R. Long-term preservation of mouse spermatozoa after freeze-drying and freezing without cryoprotection. Biol, 2003. 40. Arav A and Saragusty J. Directional freezing of sperm and associated derived technologies. Anim Reprod Sci; 2016, S0378-4320(16):30045-8. 41. Keskintepe L and Eroglu A. Freeze-Drying of Mammalian Sperm. In: Wolkers, W F and Oldenhof, H eds. Cryopreservation and Freeze-Drying Protocols. 3 ed. New York Heidelberg Dordrecht London: Springer Humana Press. 2015;.489-97.
0 Comments
Authors: Shuyana Deba, Javier Del Río and Sara Sanz Special collaboration: Álvaro Martínez Moro  Figure 1. Metaphase II oocyte (1) Infertility affects millions of couples all around the world. In spite of the solutions to their problems reproductive technology can achieve, the efficacy is eventually limited by the number and the quality of the oocytes available from the woman. In actuality, such efficiency is determined by the ovarian reserve, the oocyte quality and the maternal age, among the most important factors (2). Diminished ovarian reserve (DOR) Since ovarian reserve defines the quantity and quality of the primordial follicle pool, diminished ovarian reserve (DOR) indicates a reduction in quantity in women of reproductive age. Consequently, it represents important cause of infertility in many couples. Moreover, DOR may be associated with low pregnancy rates and high pregnancy loss regardless of age, but further research is needed in order to fully understand its implications (3). Advanced maternal age It is well known that women’s fertility declines sharply after age 35 due to several factors, which include specific issues of reproductive organs (uterus and oviducts), general health and decreasing number and quality of oocytes over time. The oocyte pool starts to decline during foetal life and continues within the reproductive life of women. Oocyte quality also decreases as a consequence of the increased rate of aneuploidies observed with age: 74% at the age of 41–42, and up to 93% after the age of 42 (5). Advanced age is too associated with a reduction in the quality of the oocyte cytoplasm (ooplasm), which directly affects oocyte maturation (3). What are the main reasons for this reduction in ooplasm quality? Mitochondria are one of the most important organelles, which are affected in different ways (6,7): - Morphological and functional abnormalities - Mitochondrial swelling - Alterations in mitochondria's cristae - Vacuolization - Alterations of the membrane potential - Alterations of the metabolic pathways in cummulus cells, which may result in impaired mitochondria biogenesis during oogenesis. These effects are due to the higher ratio of mutation consequence of the proximity of these organelles to the respiratory chain, the inefficient repair mechanism and the exposure of histories. How these changes affect oocyte quality (8)? First of all, negative effects on chromosome segregation have been observed as a result of a decreasing ATP concentration (9,10). Additionally, defects have been found in different signalling pathways such as Ca2+ signalling, which affects fertilization and the subsequent embryo development (11). 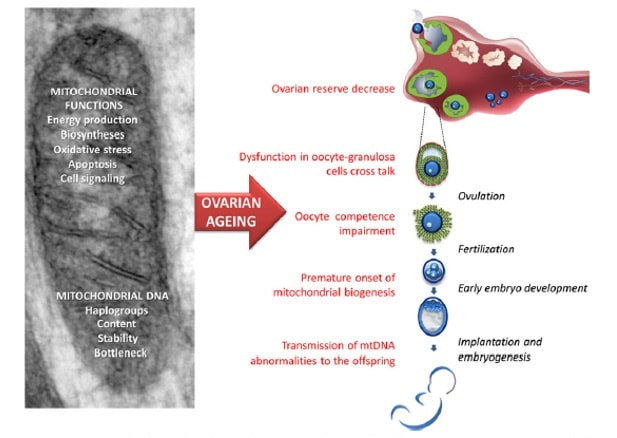 Figure 2. Overview of the multiple implications of mitochondria in ovarian ageing and their deleterious consequences on reproductive function (12) Nevertheless, different mitochondrial haplogroups should be taken into consideration. These have different bioenergetic functions, including production of reactive oxygen species (ROS) and mitochondrial coupling efficiency, aspects that might affect the oocyte longevity (13). Consequently, new techniques are being developed in order to increase the reproductive options in women with oocyte problems. Recently, one of these techniques that have been highly treated in the media is the development of additional viable oocytes from polar body genomes (2). HOW DOES TRANSFER OF POLAR BODY GENOME WORK? Originally, the transfer of polar body has been applied to cases of infertility with a genetic cause, such as the presence of mitochondrial diseases. These cases can be treated with the use of donor oocytes in clinical practice. Additionally, another application is the formation of human metaphase II (MII) oocytes, which increases the number of available oocytes for an assisted reproduction cycle (2). Two specific combined steps are needed. First, the donor oocyte spindle is removed, which requires the utilization of polarized light. Once located, it will be biopsied, obtaining an enucleated oocyte (14,15,16). Secondly, the patient polar body is biopsied, provided elimination of the spindle apparatus has been confirmed. Once both processes have been performed, the last step is the introduction of the polar body genome inside the enucleated oocyte (17). 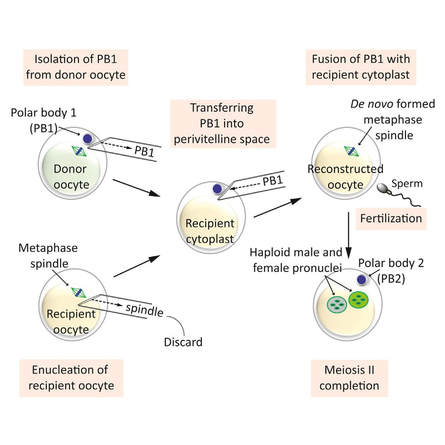 Figure 3. Polar body genome transfer technique (2) FUNCTIONAL HUMAN OOCYTES GENERATED BY TRANSFER OF POLAR BODY GENOMES Hong Ma and his group have tried to test the efficiency and possible limitations of this technique (ref). The main objective to be achieved was the formation of spindles resembling those typical of MII oocytes, including the appropriate chromosome dosage. HOW EFFICIENT IS THIS TECHNIQUE? Although DAPI staining demonstrated that all polar body nuclear transfer (PBNT)-oocytes contained spindle-chromosome complexes, only two of five experimental oocytes formed metaphase spindles similar to intact MII oocytes. This low number may be due to residual meiotic activity in enucleated human MII oocytes, which is sometimes not enough to induce formation of normal MII-like spindles. For a different cohort of oocytes, the rate of successful fertilization was 76%, still slightly lower than control oocytes. Furthermore, 42% of embryos reached blastocyst stage, indicating that most of the PBNT-oocytes were capable of completing the second meiotic division. Short tandem repeat (STR) analysis revealed that two sampled PBNT-blastocysts contained normal diploid chromosomes, determining that these embryos were completely viable. WHAT CAN BE CONCLUDED? • Polar body genome transfer seems to be a significant technique for the improvement of assisted reproductive technology (ART) outcomes and pregnancy rates, particularly for women with decreased ovarian reserve and low response to stimulation. • The cytoplasm from young donor oocytes may reduce incidences of low cytoplasmic oocyte quality. • It could provide an additional technique to support mitochondrial replacement therapy. Nevertheless, this technique is not suitable for women who cannot produce mature oocytes, typical profile of ART patients. Additionally, incidences of aneuploidy resulting from errors in mitosis or in the second meiotic division may still occur because of women advanced age. Larger datasets from this technique are needed to confirm its efficacy and safety. Also, improving preimplantation genetic screening (PGS) is critical before eventual clinical application. REFERENCES:
Authors: Shuyana Deba, Javier Del Río and Sara Sanz  Figure 1: MII oocytes obtained in vitro (1) Some weeks ago, Dr. Hayashi's group published a study in Nature in which they described how to generate functional mouse eggs from cultured stem cells. To better understand this experiment, it is necessary to explain the natural ovarian cycle. Although the experiment was carried out in mice, we believe these experiments could be of great importance in humans in the near future. Therefore, the following description applies to human ovarian cycle. GONADAL DEVELOPMENT IN THE EMBRYO As the yolk sac begins to incorporate into the embryo, germ cells migrate along the dorsal mesentery of the hindgut to the gonadal ridges, which they reach by the end of the fourth or early fifth week of development (2). Around week 7 of gestation, gonadal ridges undergo proliferation to form primitive sex cords, to which primordial germ cells (PGCs) are associated (Fig. 2). In the absence of both expression of the SRY gene product and male sex hormones, differentiation into female organs is determined (3,4). Whereas the origin of the follicular cells of the ovary remains unclear, likely candidates are cells from the coelomic epithelium and the mesonephros. The follicular cells eventually associate with the PGCs to form primordial ovarian follicles (3,4). ORIGIN OF GERM CELLS Human oocytes are derived from round PGCs that can be identified in the wall of the yolk sac as early as 24 days after fertilization. Once the gonad develops into an ovary, PGCs become oogonia, and mitotic divisions go on up to the seventh foetal month, and cease at some point shortly before birth (5). By the 8-9th week after fertilization, some oogonia enter into prophase I of meiosis, thus becoming primary oocytes. Meiosis starts in the deepest part of ovary (medulla) and progresses towards the cortex. Some time after it has been incorporated into a primordial follicle, the oocyte arrests after diplotene, at a particular stage called dictyate or dictyotene. This means the oocyte enters into a long quiescence period that begins before birth and ends up either resuming after the LH surge (leading to ovulation) or in follicular atresia (3). Even though the experiment by Hayashi's group has just been successfully performed in mice, the purpose of the investigation is eventually applying it to humans. 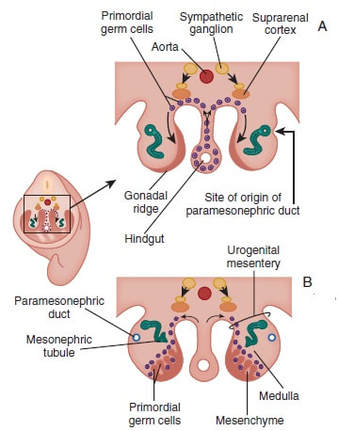 Figure 2: Gonadal development. C: Transverse sections showing the gonadal ridges and the migration of primordial germ cells into the developing gonads; D: transverse section through a 6-week-old embryo, showing the primary sex cords and the developing paramesonephric ducts (3) FOLLICULOGENESIS AFTER BIRTH Once placental oestrogen disappears, gonadotrophin levels begin to increase after birth. This increment will last 12-24 months postpartum in women. Thus, the ovaries remain inactive until the beginning of pubertal stage, moment when they will start showing response to GnRH pulsatility (6). By this time, ovaries contains germ cells at the primordial follicle phase, which are composed of small immature oocytes arrested at dictyate, and surrounded by a single layer of flat squamous granulosa cells (7). WHAT HAPPENS AFTER PUBERTY? Once sexual maturity is reached, multiple primordial follicles are activated from their quiescent state, resuming folliculogenesis by cohorts every menstrual cycle. During follicle maturation, follicles are subjected to activin, AMH, FSH, inhibin, LH, TGFβ and a multitude of other molecules that control the process. At the beginning of this process, as the oocyte resumes meiosis follicular cells proliferate and surround the oocyte, which develops into a primary follicle (7,8,9). Briefly, transition into the secondary or antral follicle stage involves the alignment of the stroma around the basal lamina and the development of an independent blood supply. Follicular cells of the underlying stroma, the granulosa cells, subsequently differentiate into an inner and an outer theca layers, the latter supporting this blood supply. The production of follicular fluid by these cells promotes the formation of the antrum in pre-antral follicles (10). Progression through antral follicle development is promoted by FSH and coincides with (7): - Completion of oocyte growth. - Acquisition of competence to complete meiosis (dependent on fertilization). - Continued granulosa cell proliferation. - Differentiation of cummulus and mural granulosa cells upon formation of the follicular antrum. Response to FSH causes growth of the follicle by proliferation of follicular cells and coalescence of the astral cavities, thus generating a follicle whose volume keeps increasing. The final stage of folliculogenesis takes place when the Graafian follicle reaches its maximum size (around 20 mm diameter). The LH surge triggers the ovulation of a metaphase II-arrested oocyte (along with the cummulus), capable of being fertilized and able to support embryonic development (7). Although there are notable differences between species, in humans, usually only one follicle is capable of completing growth and maturation up to ovulation (preovulatory), due to its more efficient response to FSH in a "competitive manner" against the rest of developing follicles. The one follicle will progress until subsequent fertilization (dominant follicle), whereas the remaining follicles in the cohort will undergo atresia (8). 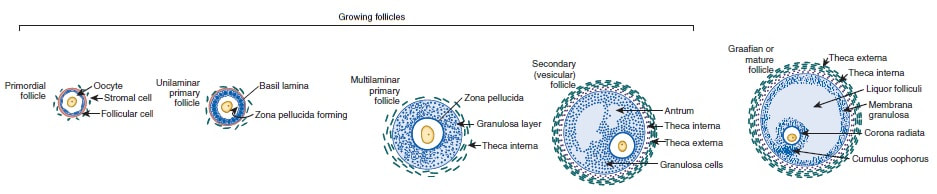 Figure 3. Shematic drawing of development of ovarian follicles (3) ABOUT PLURIPOTENT STEM CELLS For centuries, scientists have investigated how oocytes and sperm develop in mammals. Most of this work has been performed in non-human species, mainly the mouse. Human pluripotent stem cells (PSCs)-derived gamete research presents notable scientific value and potential, both for understanding basic mechanisms of gamete biology and for overcoming clinical problems (11). PSCs are currently used to answer important scientific questions such as the role of specific genes in early germ cell development, involved pathways, interaction between germ cells and supporting somatic cells, or partly or entirely in vitro PSCs-derived eggs and sperm (11). Some of the potential benefits of these new techniques could be (11): - Additional options for assisted human reproduction. - New ways to prevent and treat infertility, genetic diseases and some types of cancer. - Optimization of the number of oocytes obtained from women. "IN VITRO RECONSTITUTION OF THE ENTIRE CYCLE OF THE MOUSE FEMALE GERMLINE" (1) The authors have shown a method to reconstruct the entire process of mouse oogenesis in vitro, using embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs) in order to obtain fully functional and mature oocytes. Why does this study mean so much? First of all, this is a big achievement in the field of regenerative medicine, through which it is possible to study the function of certain genes involved in oogenesis. Furthermore, it sets the ground for future similar studies and culture systems in other specie, and as such, it may result in a great advance in reproductive medicine. However, which are the limitations for this approach? The culture system requires somatic cells, which are obtained from embryos. So far, this is an ethical and legal issue in humans. A feasible solution would be to obtain the gonadal somatic cell-like cells from PSCs. The authors found that the differentially expressed genes (DEGs) were down-regulated in in vitro MII oocytes, whereas in vivo MII oocytes showed an increase in the number of transcripts. This maybe a consequence to the fact that, during in vitro growth (IVG) and in vitro maturation (IVM), oocyte development was jeopardized within a subset of (or perhaps all) the oocytes. Why? Misregulation of such genes, related to mitochondrial functions, might attenuate the potential of the in vitro MII oocytes. This problem could be solved by an analysis of the metabolic pathway, which could in turn provide information to refine both IVG and IVM. RESULTS, "PROBLEMS" AND "ACHIEVEMENTS" OF THE EXPERIMENTS (1)  As it can be seen concluded from above, it seems clear that further tests are still needed, which would help measure some aspects of the viability and functionality of this in vitro generated gametes.
REFERENCES 1. Hikabe O, Hamazaki N, Nagamatsu G, Obata Y, Hirao Y, Hamada N et al. Reconstitution in vitro of the entire cycle of the mouse female germ line. Nature. 2016;. 2. Baillet A, Mandon-Pepin B. Mammalian ovary differentiation–A focus on female meiosis. Molecular and Cellular Endocrinology. 2012;356(1-2):13-23. 3. Bolender DL, Kaplan S. Chapter 3 – Basic Embryology. In: Fetal and Neonatal Physiology. 2017. p. 23–39.e2. 4. Parker KL, Schimmer BP. Chapter 8 – Embryology and Genetics of the Mammalian Gonads and Ducts. In: Knobil and Neill’s Physiology of Reproduction. 2006. p. 313–36. 5. Racowsky C, Gelety TJ. Chapter 4 – The Biology of the ovary. Princ Med Biol. 1998;12:77–102. 6. David Vantman B, Margarita Vega B. Fisiología reproductiva y cambios evolutivos con la edad de la mujer. Revista Médica Clínica Las Condes. 2010;21(3):348-362. 7. Hutt K, Albertini D. An oocentric view of folliculogenesis and embryogenesis. Reproductive BioMedicine Online. 2007;14(6):758-764. 8. Green L, Shikanov A. In vitro culture methods of preantral follicles. Theriogenology. 2016;86(1):229-238. 9. Palermo R. Differential actions of FSH and LH during folliculogenesis. Reproductive BioMedicine Online. 2007;15(3):326-337. 10. Gougeon A. Human ovarian follicular development: From activation of resting follicles to preovulatory maturation. Annales d'Endocrinologie. 2010;71(3):132-143. 11. Mathews D, Donovan P, Harris J, Lovell-Badge R, Savulescu J, Faden R. Pluripotent Stem Cell-Derived Gametes: Truth and (Potential) Consequences. Cell Stem Cell. 2009;5(1):11-14. |
Entries
March 2019
Categories
All
 2016-2019. All Rights Reserved by Embryologist Media. This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License . |
Embryologist Media
