|
Authors: María Caballero Sastre & Raquel Pillado González “Currently, performing any cryopreservation technique results in some degree of damage to the sperm.“ 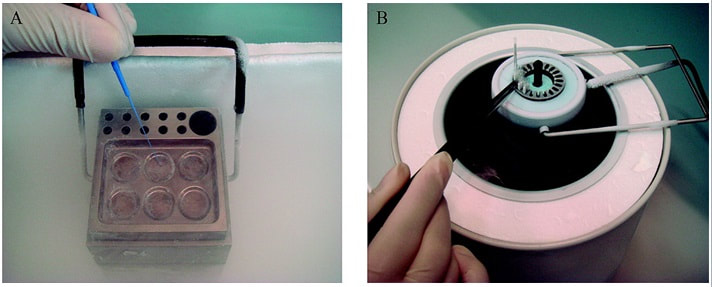 Figure 1. Solid Surface Vitrification (A) and slow cooling equipment (B) (1). THE HISTORY OF SPERM CRYOPRESERVATION Human semen cryopreservation has a long history that begins at the end of the nineteenth century. After prior observations on sperm surviving cooling at very low temperatures (-150º C) (2), Mantegazza (1866) first suggested the idea of human sperm banks (3). Years later, Mantegazza proved it was possible to extend human sperm lifespan up to four days by cooling at moderate sub-zero temperatures (-17º C) (4). Although a few extra days of storability does not make much difference in practical terms, this was the starting point of further research aimed to develop techniques that would allow for longer storage periods. The next leap forward in this field was the discovery of the cryoprotectant properties of glycerol in 1949. This molecule proved to be an effective cryoprotectant agent (CPA) when combined with bovine sperm, and allowed for the development of new cryopreservation methods through which sperm could preserve their motility and fertilization capabilities even after the freezing-thawing process (5). However, during the following years this new technique was primarily applied to cryopreservation of farm animal semen rather than human semen (6). In 1953, at the University of Iowa, the first case of live birth from cryopreserved sperm was reported (7). This successful birth, along with reports of high survival percentage (67%) of human sperm after cryopreservation, popularized the utilization of glycerol with human semen. The most common method to use glycerol was the protocol described by Bunge and colleagues, which stated the processing of sperm in a 10%-glycerol solution before freezing it with dry ice (7). A decade later, the use of liquid nitrogen was introduced for long-term sperm cryopreservation. This new method led to the progressive normalization of sperm freezing as a widespread practice in healthcare (8). With the availability of long-term storage and the extended use of sperm freezing, new methods and variants were developed over time, such as slow freezing, fast freezing, LN2 vapours or lyophilization, which will be later described. Nowadays, cryopreservation is routinely used in most assisted reproduction centres for numerous reasons:
It is important to possess a clear understanding of the semen cryopreservation process due to its current importance in clinical and research environments. Modern assisted reproduction practices are unthinkable without this tool. CRYOPRESERVATION TECHNIQUES Before being frozen, a sperm sample needs to be appropriately processed in order to separate sperm cells from the seminal plasma. This helps increase the concentration of high quality spermatozoa for a later use. Different techniques for sperm selection have been reviewed in our previous post. Cryoprotectants The process of cryopreservation may involve irreversible cellular damage due to a change in the osmotic balance (11). Upon freezing of the extracellular water, the solute concentration increases in this fraction. As a result, the intracellular water is transported out of the cell to restore the osmotic balance, which may lead to cell dehydration and irreversible membrane damage (12, 13). Consequently, CPAs need to be incorporated along the sperm sample following processing. These molecules will protect spermatozoa by reducing intracellular ice formation and by decreasing the freezing point of the medium and the solute concentration present in the extracellular environment (14). There are two kinds of cryoprotective agents: permeable and non-permeable. The permeable ones, such as glycerol, dimethyl sulfoxide or ethylene glycol, protect the intracellular structures and biomolecules. Non-permeable agents, like sucrose, polysaccharides and some proteins, contribute to keeping the osmotic equilibrium, thus preventing cellular dehydration (11). Procedures - Cryopreservation in liquid nitrogen (LN2) This is the most commonly used method to cryopreserve sperm. It can be subdivided into three methods:
- Cryopreservation in microdroplets Microdroplets are sometimes used in the clinic to cryopreserve sperm in small volumes. This is preferred in cases such as epididymal sperm aspiration. Around 50-100 μL of the sperm-CPA solution are placed on a dry ice plate up to freezing (Fig. 2); droplets formed in such a way are then kept in vials and plunged into LN2 (18).  Figure 2. Cryopreservation in droplets. Dry ice plate used to cool down samples that form sperm droplets (in yellow). - Vitrification This technique consists of cooling the sample at ultrarapid rates, so that the water solidifies (vitrifies) as a glass-like structure rather than forming ice crystals (19). However, even though vitrification should cause minimal damage, this is not always the case in clinical practice. One of the most frequent problems is the requirement for very high CPA concentrations that sperm do not tolerate well (20). Despite this, some studies have demonstrated it is possible to perform vitrification without using CPAs (20-22). Also, the large volume of sperm typically used impedes the cooling of the sample at the appropriate speed, causing ice formation (4). Despite these limitations, a vast proportion of clinics use vitrification as a routine practice due to its practical advantages. In recent years, a new variant of this technique has been developed. This evolved version is called Solid Surface Vitrification (SSV) (23, 24). For this procedure, the sample or tissue is directly exposed to a metal surface previously precooled at -160º C before the use of LN2 (Fig. 1). This method prevents the apparition of nitrogen bubbles and evaporation that would slow the cooling rate (24). This technique has been previously applied to animal mature oocytes and human gametes and embryos, yielding successful results (23, 24). OUTCOME COMPARATIVE BETWEEN TECHNIQUES Because every cryopreservation technique shows different advantages and disadvantages, it is important to bear in mind how sperm will be affected during the thawing process. Parameters such as motility, viability, morphology and DNA integrity are evaluated accordingly. Regarding LN2 techniques, no studies so far have directly compared post-thaw sperm quality following slow and rapid freezing. The literature shows agreement between studies on the main problem in both processes, which is controlling cooling rates (reviewed in 16). If the cooling rate is too fast, ice crystals may be formed inside the cells. By contrast, if it is too slow, the result could be cell contraction due to osmotic stress (25). This issue can be faced by using an automated programmable freezer, but only when keeping a large number of samples (26). Even though several reports have compared fast and slow freezing in animal reproduction, conclusions are controversial. Some studies in semen from horse (27) and buffalo (28) have reported better results when using fast thawing, whereas other authors have found no difference between fast and slow thawing rates (29). Nevertheless, it seems clear that the critical point lays on thawing matching the freezing process. Considering rapid freezing, thawing is recommended to be also carried out at a fast rate to avoid formation of intracellular ice crystals. Likewise, for the slow-rate cooling procedure, the sample needs a slow thawing protocol, since cells need more time to rehydrate (30). On the other hand, all studies on the use of LN2 show similar or slightly better results regarding the aforementioned parameters when using nitrogen vapours (31-33). These results, however, are obtained after short-term storage of samples in nitrogen vapours of up to three months. When stored for longer, sperm quality decreases. Consequently, this method is only recommended for short-term storage (further research would be needed in order to support its application for long-term storage) (34). Upon comparison between vitrification and LN2 techniques, different results can be highlighted. Certain authors determined that results of sperm parameters such as motility, viability and normal morphology were similar between vitrification and rapid freezing techniques (21). On the contrary, different results were found for DNA fragmentation rates. Whilst some groups found that DNA fragmentation was significantly higher for the rapid freezing technique (12, 21) or for LN2 vapours (24), other groups obtained contradictory or uncertain results (35, 36). For instance, DNA fragmentation has been observed to increase over time when analyzing semen 6h after thawing, compared to recently-thawed samples (35). Despite the different results obtained, vitrification shows important advantages compared to other available techniques. Some of these advantages are: 1) unnecessary use of CPAs; 2) the technique is simpler and faster compared to conventional slow freezing, due to the fact that once the sample is kept in a proper container (such as cryoloop or straws), it is rapidly plunging into LN2 to be stored; 3) no requirement for programmable freezers; 4) the sample is free of seminal plasma and potential pathogens (vitrification is usually performed after swim-up); 5) no requirement for post-thaw processing (reviewed in 6). TRENDS IN IMPROVING SEMEN CRYOPRESERVATION Currently, performing any cryopreservation technique results in some degree of damage to the sperm (4). The severity may differ depending on the initial quality of the sample, being greater in poor quality semen. Luckily, the application of ICSI allows for the successful use of low quality sperm (if necessary) even after having been cryopreserved. Side effects of cryopreservation on sperm include reduced motility, vitality, viability and increased DNA damage. Although motility is the most affected parameter, DNA damage entails greater detrimental effects regarding embryo viability (6). The majority of the harm produced by cryopreservation occurs during the freezing and thawing phases, the crucial moments being between -15º C and -60º C. It is worth mentioning that a considerable damage is produced by CPAs themselves; these agents cause oxidative stress that derives in the formation of reactive oxygen species (ROS) (8, 10, 37). Furthermore, these components affect the polyunsaturated fatty acids in plasmatic membranes due to lipid peroxidation (10). Consequently, phospholipids reservoirs such as egg yolk are usually added to the freezing media. As a more direct countermeasure, several current research lines look into numerous antioxidant components in order to be added to freezing media, thus avoiding the damage caused by ROS. Examples of these antioxidants are TAT-peroxiredoxin-2 fusion protein, quercetin or melatonin. Preliminary research indicates that their presence results in higher motility and viability rates post-thawing, along with a reduction of intracellular ROS levels (8, 10). Other approaches consider the utilization of protocols that may directly disregard the use of CPAs, such as certain vitrification protocols previously mentioned. Sperm freezing entails other associated problems, too, such as the loss of chromatin and acrosome integrity that had been observed post-thaw. Recent data have reported differences in the levels of DNA and acrosome integrity after cryopreservation depending on the freezing technique used (24). The difference in DNA integrity levels is suspected to be due to the cold shock faced by the samples (24). This issue could be amended by the development of media able to preserve sperm without freezing, an avenue that is currently being pursued. For instance, Riel and colleagues have reported that the use of an electrolyte-free medium for short-time (1 week) storage of semen yields better levels of DNA integrity in comparison to traditional cryopreservation. If the storage period capacity could be further improved, this might become a rather attractive alternative (38). Lyophilization or freeze drying is an experimental technique that has been proven less harmful to the DNA (4, 10, 38). In order to perform this method, the sample must be cooled below the triple point of water (Fig. 3). At this temperature solid water (ice) sublimates when the pressure is decreased and exits the cell, leaving it fully dehydrated (4, 10). However, this process irreversibly damages the sperm membrane, thus resulting in non-motile or even non-viable (dead) sperm. Nevertheless, studies on mouse sperm have shown that lyophilized spermatozoa can be used for fertilization with the assistance of ICSI (39). Although the first attempts to use lyophilization on human sperm were in the 50s, today there is still a lacking protocol for this technique that is able to preserve both sperm motility and viability (4). 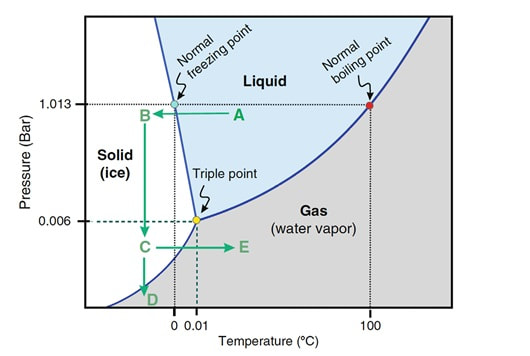 Figure 3. Water phase diagram showing the relation of the conditions of temperature and pressure for freeze-drying (not in scale). Samples are frozen by reducing the temperature (A to B) and then the pressure is also reduced by aspiration (creating vacuum) so the sample lies below the ‘triple’ point (C) for both temperature and pressure (this is the point where all states co-exist). From here on the sample is subjected to a controlled increase of temperature or to a further decrease of pressure to sublimate ice (for detailed current sperm freeze-drying protocols, see (40) and (41)). Modified from (41). The main advantages of freeze drying are: the possibility to preserve spermatozoa with high DNA integrity for at least a year and a half (39), the inactivation of viruses that may be present and the fact that liquid nitrogen is not required. Additionally, samples can be stored at 4º C and transported at room temperature (4). To date, this method still remains experimental regarding humans, due to the lack of actual data on the matter (4, 10). CONCLUSIONS Cryopreservation has gone a long way. Its use in reproductive medicine got to revolutionize the horizon for infertile couples. New doors opened decades ago, and it is fair to reason new ones will open in the near future. Egg donation, social freezing, embryo cryopreservation. Times and timing have changed for patients, and clinics and reproduction centres faced the need for evolution in order to cope with rising approaches. In spite of the variety of options for semen cryopreservation, all of them present their own limitations. Continuous research allows for the discovery of new ways to correct these flaws; however, there lies a long path ahead, and further studies will be required before any improvement can be incorporated to routine practice. REFERENCES
1. Wang X, Catt S, Pangestu M and Temple-Smith P. Live offspring from vitrified blastocysts derived from fresh and cryopreserved ovarian tissue grafts of adult mice. Soc Reprod Fert.2009;138(3): 527–535. 2. Varghese AC, Nandi P, Mahfouz R, Athayde KS, Agarwal A. Human Sperm Cryopreservation. In: Varghese, AC., Nandi, P., Mahfouz, R., Athayde, KS., Agarwal A, editor. ANDROLOGY LABORATORY MANUAL [Internet]. Cleveland Clinic. 2014; p.196–206. 3. Bunge, GR., & Sherman, KJ. Fertilizing Capacity of Frozen Human Spermatozoa. Nature.1953; 172(4382):767-8. 4. Mocé E, Fajardo AJ, Graham JK. Human sperm cryopreservation. EMJ. 2016;1:86–91. 5. Polge C, Smith AV, and Parkes AS. Revival of Spermatozoa after Vitrification and Dehydration at Low Temperatures Nature.1949; 164, 666. 6. Sharma R, Kattoor AJ, Ghulmiyyah J, Agarwal A, Sharma R, Kattoor AJ, et al. Effect of sperm storage and selection techniques on sperm parameters. Syst Biol Reprod Med. 2015;61(1):1–12. 7. Bunge RG, Sherman JK. Fertilizing capacity of frozen human spermatozoa. Nature. 1953;172:767–768. doi: 10.1038/172767b0. 8. Rozati H, Handley T, Jayasena C. Process and Pitfalls of Sperm Cryopreservation. J Clin Med. 2017;6(89):1–13. 9. Tiwari A, Tekcan M, Sati L, Murk W, Stronk J. A new media without animal component for sperm cryopreservation : motility and various attributes affecting paternal contribution of sperm. J Assist Reprod Genet. Journal of Assisted Reproduction and Genetics; 2017;34:647–57. 10. Karimfar MH, Niazvand F, Haghani K, Ghafourian S, Shirazi R, Bakhtiyari S. The protective effects of melatonin against cryopreservation-induced oxidative stress in human sperm. Int J Immunopathol Pharmacol. 2015;28(1):69–76. 11. Sieme H, Oldenhof H, Wolkers WF. Mode of action of cryoprotectants for sperm preservation. Anim Reprod Sci [Internet]. Elsevier B.V.; 2016;1–14. 12. Elliott GD, Wang S, Fuller BJ. Cryobiology Cryoprotectants : A review of the actions and applications of cryoprotective solutes that modulate cell recovery from ultra-low temperatures. Cryobiology [Internet]. Elsevier Ltd; 2017;76:74–91. 13. Royere D, Barthelemy C, Hamamah S, Lansac J. Cryopreservation of spermatozoa: a 1996 review. Hum Reprod Update. 1996;2(6):553-9. 14. Thachil JV, and Jewett, MA. Preservation techniques for human semen. Fertil Steril. 1981;35:546–8. 15. Holt, WV. Basic aspects of frozen storage of semen. Anim Reprod Sci. 2000; 62(1–3):3-22. 16. Sherman, JK. Cryopreservation of human semen. In: Handbook of the Laboratory Diagnosis and Treatment of Infertility. 1990; Keel B. and Webster BW, eds. Boca Raton, Fla, USA: CRC Press, pp. 229–59. 17. Fountain D, Ralston M, Higgins N, Gorlin, J, Uhl L, Wheeler C, et al. Liquid nitrogen freezers: a potential source of microbial contamination of hematopoietic stem cell components. Transfusion, 1997;37:585–91. 18. Abdelhafez F, Mohamed B, El-nashar S, Sabanegh E, Desai N. Techniques for cryopreservation of individual or small numbers of human spermatozoa : a systematic review. Hum Reprod. 2018;15(2):153–64. 19. Kuleshova LL, Lopata A. Vitrification can be more favorable than slow cooling. Fertil Steril. 2002;78(3):449-54. 20. Isachenko E, Isachenko V, Katkov II, Dessole S, Nawroth F. Vitrification of mammalian spermatozoa in the absence of cryoprotectants: from past practical difficulties to present success. Reprod Biomed Online. 2003;6(2):191-200. 21. Agha-Rahimi A, Khalili MA, Nabi A, Ashourzadeh S. Vitrification is not superior to rapid freezing of normozoospermic spermatozoa: effects on sperm parameters, DNA fragmentation and hyaluronan binding. Reprod Biomed Online. 2014;28(3):352-8. 22. Nawroth F, Isachenko V, Dessole S, Rahimi G, Farina M, Vargiu N et al. Vitrification of human spermatozoa without cryoprotectants. Cryo Letters. 2002;23:93–102. 23. Kamath, MS., Muthukumar, K., Appendix B: Solid Surface Vitrification. Methods Mol. Biol.2017;1568:297-307. 24. Rahiminia T, Hosseini A, Anvari M, Ghasemi-esmailabad S, Talebi AR. Modern human sperm freezing: Effect on DNA , chromatin and acrosome integrity. Taiwan J Obs Gynecol. 2017;56(Feb):472–6. 25. Said TM, Gaglani A, Agarwal A. Implication of apoptosis in sperm cryoinjury. Reprod Biomed Online. 2010;21(4):456-62. 26. Pugliesi G, Fürst R, Carvalho GR. Impact of using a fast-freezing technique and different thawing protocols on viability and fertility of frozen equine spermatozoa. Andrologia. 2014;46(9):1055-62. 27. Fürst R, Carvalho GR, Fürst MCO, Ruas JRM, Borges AM, et al. Efeito do resfriamento do sêmen eqüino sobre sua congelabilidade. Arq Bras Vet Zootec. 2005;57:599–607. 28. Shah SA, Andrabi SM, Qureshi IZ. Effect of equilibration times, freezing, and thawing rates on post-thaw quality of buffalo (Bubalus bubalis) bull spermatozoa. Andrology. 2016;4(5):972-6.b. 29. Vidament M, Yvon JM, Couty I, Arnaud G, Nguekam- Feugang J, et al. Advances in cryopreservation in modified INRA 82. Anim Reprod Sci 68:201–218. 30. Mazur P. Basic concepts in freezing cells. In: Proc. 1st International Conf. Deep Freezing Boar Semen. Uppsala, Sweden, 2005;91–111. 31. Amesse LS, Srivastava G, Uddin D, and Pfaff-Amesse T. Comparison of cryopreserved sperm in vaporous and liquid nitrogen. J Reprod Med. 2003;48:319–24. 32. Saritha KR, and Bongso, A. Comparative evaluation of fresh and washed human sperm cryopreserved in vapor and liquid phases of liquid nitrogen. J Androl. 2001;22:857–62. 33. Satirapod C, Treetampinich C, Weerakiet S, Wongkularb A, Rattanasiri S, et al. Comparison of cryopreserved human sperm from solid surface vitrification and standard vapor freezing method: on motility, morphology, vitality and DNA integrity. Andrologia. 2012;44(Suppl. 1):786–790. 34. Lim JJ, Shin TE, Song S, Bak CW, Yoon TK and Lee DR. Effect of liquid nitrogen vapor storage on the motility, viability, morphology, deoxyribonucleic acid integrity, and mitochondrial potential of frozen-thawed human spermatozoa. Fertil Steril. 2010;94:2736–41. 35. Gosalvez J, Nunez R, Fernandez JL, Lopez-Fernandez C, Caballero P. Dynamics of sperm DNA damage in fresh versus frozen–thawed and gradient processed ejaculates in human donors. Andrologia- 2011;43:373–377. 36. Isachenko E, Isachenko V, Katkov II, Rahimi G, Schondorf T, et al. DNA integrity and motility of human spermatozoa after standard slow freezing versus cryoprotectant-free vitrification. Hum. Reprod.2004;19:932–939. 37. Wu Y. Successful delivery derived from cryopreserved rare human spermatozoa with novel cryopiece. Am Soc Androl. 2017;5:832–7. 38. Gianaroli L et al. DNA integrity is maintained after freeze-drying of human spermatozoa. Fertil Steril. 2012;97(5):1067-73. 39. Ward MA, Kaneko T, Kusakabe H, Biggers JD, Whittingham DG and Yanagimachi R. Long-term preservation of mouse spermatozoa after freeze-drying and freezing without cryoprotection. Biol, 2003. 40. Arav A and Saragusty J. Directional freezing of sperm and associated derived technologies. Anim Reprod Sci; 2016, S0378-4320(16):30045-8. 41. Keskintepe L and Eroglu A. Freeze-Drying of Mammalian Sperm. In: Wolkers, W F and Oldenhof, H eds. Cryopreservation and Freeze-Drying Protocols. 3 ed. New York Heidelberg Dordrecht London: Springer Humana Press. 2015;.489-97.
0 Comments
Authors: Iris Martínez Rodero and Raquel Pillado González "Selection of the best spermatozoon for ICSI is critical and it is directly linked to fertilization rates, optimal embryo development and implantation rates" 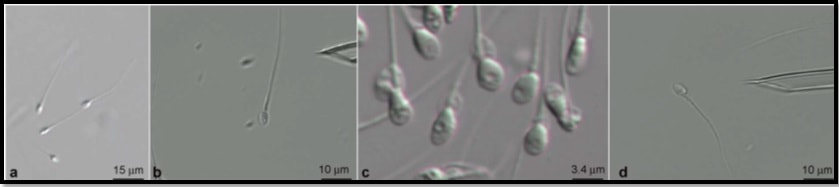 Figure 1. Spermatozoa observed at low magnification (a) and high magnification (b–d). The shape and presence of vacuoles can be clearly observed in (c). A vacuole-free spermatozoon is shown in (d). INTRODUCTION It is estimated that approximately 10% of couples seek help to overcome infertility problems. Data from the American Society of Reproductive Medicine points to male factor as the reason behind one third of couples’ infertility problems (1). The remaining two thirds seem to be equally distributed between female-related problems and a mixture of unexplained causes and combinations of male and female factors, respectively (1). As continuation of our previous post on sperm selection, we revisit the topic from the perspective of the numerous techniques currently available to be specifically used prior intracytoplasmic sperm injection (ICSI). This procedure is the advised treatment for most cases of male factor infertility; in fact, ICSI accounts for 70-80% of cycles performed globally (2). ICSI candidates often present low sperm count and/or increased probability of genetic abnormalities, compromised DNA and morphological defects of spermatozoa. Therefore, selection of the best spermatozoon for ICSI is critical and it is directly linked to fertilization rates, optimal embryo development and implantation rates (3). Traditional sperm selection procedures (swim-up and density gradient centrifugation (DGC)) are based just on motility rather than molecular features. Consequently, in order to choose the best spermatozoa, researchers have developed different non-invasive methods to process and select sperm for ICSI (Table 1) (2). 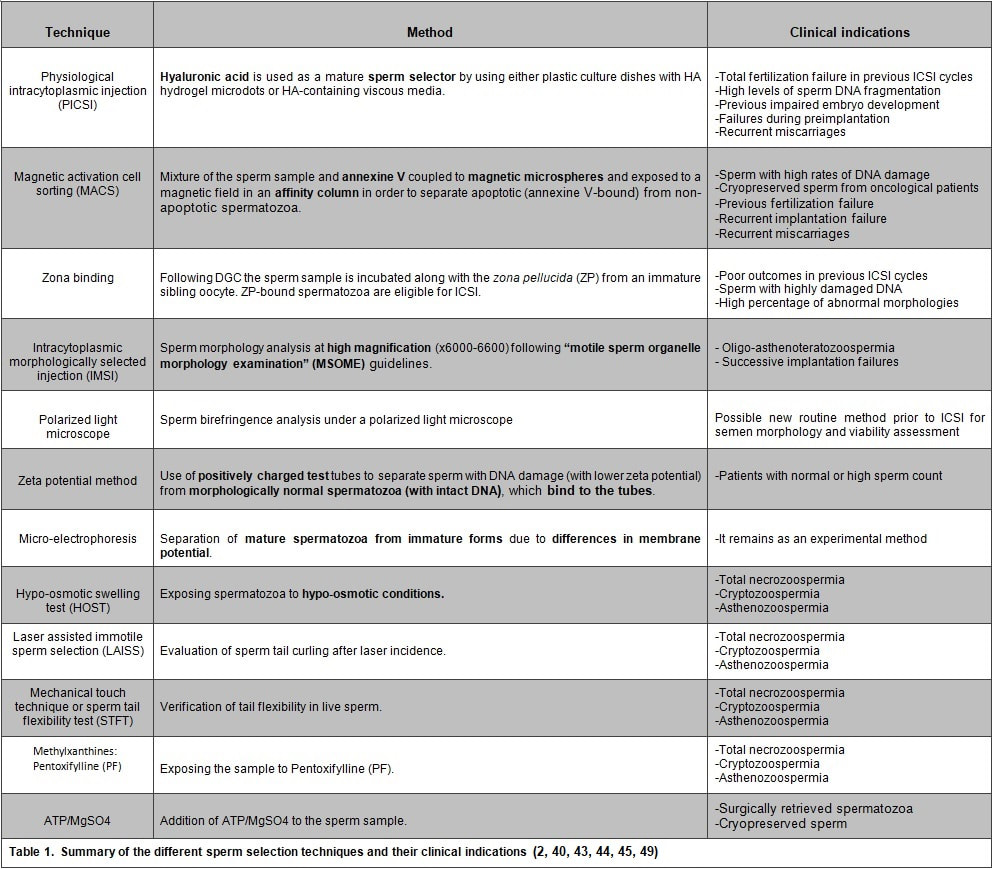 SPERM SELECTION TECHNIQUES Sperm selection prior to fertilization is a crucial step for IVF success, especially in cases of low number of available oocytes to inject (4). Several techniques are currently utilised for this purpose, each of them relying on different features in order to select the optimal sperm. BINDING ASSAY TECHNIQUES Physiological intracytoplasmic injection (PICSI) This technique is based on the fact that hyaluronic acid (HA) plays an important role in selecting functionally competent sperm during in vivo fertilization (5). Not only HA is the main component of the cumulus matrix that surrounds the human oocyte, but also a natural selector of developmentally mature sperm. Furthermore, experimental data have shown that it can be used with a similar effect in vitro. HA-binding spermatozoa in vitro exhibit complete plasma membrane remodelling, decreased levels of cytoplasmic inclusions and residual histones, nuclear maturation and reduced risk of chromosome imbalance (i.e. chromosomal aneuploidy) and chromatin diseases (4). Currently, there are two options available to perform this technique. First, plastic culture dishes with HA hydrogel microdots attached to the bottom. After using this method the desired spermatozoa remain bound to the microdots by the head, while flagella beat in place (4). Secondly, the use of an alternate HA-containing viscous medium, method known as “Sperm Slow” because the spermatozoa appear “slowed down” (4). Since PICSI is considered to better assess the physiological potential in order to select sperm for ICSI, some authors claim it should replace the use of conventional ICSI and potentially toxic polyvinylpyrrolidone (PVP) (used to slow down sperm movement). Nevertheless, significant improvement of IVF outcomes using HA-based sperm selection is still to be confirmed in larger studies (6, 7), reason why the use of PICSI is currently limited to punctual cases in which significant improvement of clinical outcomes has been proved (8). Such cases include total fertilization failure by ICSI, high levels of DNA fragmentation, impaired embryo development, failure to implant in the uterus and recurrent miscarriages (9, 10). Fertilization failure after ICSI can sometimes be explained by problems in sperm nucleus decondensation and chromatin abnormalities related to DNA fragmentation (11). Sperm with such DNA damage and/or chromosome unbalance are avoided by PICSI/HA binding negative selection (3). Although it is a controversial issue, several authors have found a significant improvement of embryo quality and implantation ability when performing PICSI vs conventional ICSI, as well as a reduced number of miscarriages (3, 11, 12, 13, 14). The decreased incidence of miscarriages seems to be linked to the incapacity of sperm with genetic defects to form a normal pro-nucleus after conventional ICSI, thus generating aneuploid embryos with high levels of fragmentation during cleavage. Such aberrant embryos normally end up in pregnancy loss (14). Since PICSI allows for selection of sperm with properly compacted chromatin, embryos derived from these spermatozoa are less likely to present defects in quality (15). This translates into a higher implantation potential and a lower incidence of miscarriage, which eventually improves the reproductive outcome (12, 13). Magnetic activation cell sorting (MACS) In 2008 Said and colleagues proposed a protocol combining a new method, denominated magnetic activation cell sorting (MACS), and density gradient column (DGC) in order to obtain higher-quality sperm samples (2). MACS consists in mixing up the semen sample with annexine V (A5) (known to have high affinity with phosphatidylserine (PS)) coupled to magnetic microspheres. The sample is then exposed to a magnetic field in an affinity column (16). A5-bound sperm have been linked to PS externalisation, a well-known sign of apoptosis due to the loss of membrane integrity. MACS should be performed prior to DGC because PS externalisation naturally occurs during sperm capacitation by DGC, and so MACS may inadvertently discard good-quality sperm (17). During the last decade, several studies have corroborated the efficient use of this protocol in order to obtain sperm samples with a low DNA fragmentation index and higher fertilization potential (2, 16). Because MACS reduces the percentage of apoptotic sperm selected from the sample, this technique is mostly indicated in cases of high rates of DNA damage or predisposition to suffer from it (18). Such cases include: patients with altered sperm parameters, cryopreserved spermatozoa from cancer patients (19), previous fertilization failure, recurrent implantation failure and recurrent miscarriage (20, 21). Several clinics have implemented MACS in their daily routine, claiming selection of sperm through MACS to improve their ICSI outcome (22, 23, 24). However, such statement remains controversial, since other authors have reported no actual beneficial effect after using this technique (16, 25). Zona binding This method mimics natural sperm selection using the zona pellucida (ZP) from an immature sibling oocyte (26). The sperm sample is processed by DGC and then incubated along with the ZP for two hours. ZP-bound sperm are then eligible for ICSI. Although various studies have linked the ZP binding assay prior to ICSI to improved embryo quality and implantation rate, it is still not feasible to employ this method on a routine basis due to the extra amount of work and time required. Therefore, it is currently recommended to remain restricted to patients with poor outcomes in previous ICSI cycles, or to those who exhibit sperm with damaged DNA or abnormal morphologies (2). MICROSCOPY BASED TECHNIQUES Intracytoplasmic morphologically selected injection (IMSI) Studies have shown that sperm classified as morphologically “normal” at conventional optical resolution and magnification (x200-x400) may carry ultrastructural defects. Many of these abnormalities have been linked to hidden chromosomal defects, high levels of DNA fragmentation, abnormal centriolar function, etc. (1, 2). These aberrations may impede fertilization, lead to post-fertilization arrest or even disrupt embryo development (2). IMSI is an approach developed from a method of sperm evaluation, first described in 2002 by Bartoov and colleagues and denominated “motile sperm organelle morphology examination” (MSOME) (27). In order to apply this evaluation technique, observations need to be performed at x6000-6600 magnification (1). Performance of sperm selection along with MSOME requires the use of specific equipment. This should be an inverted light microscope with high-power optics intended for differential interference contrast and enhanced by digital imaging, which allows the embryologist to assess sperm morphology in real time. Sperm selection is carried out in a dish different from the one used for ICSI; it is a glass-bottomed dish that allows for the best optimal quality. Semen samples for IMSI are subjected to discontinuous DGC beforehand, but the exact procedure may vary depending on the quality of the sample (2). Sperm selection based on IMSI—MSOME findings helps to discard spermatozoa with mitochondrial dysfunction or DNA damage (4). In this regard, IMSI-MSOME has been proved useful for oligo-asthenoteratozoospermic patients (28) and for couples with recurrent implantation failures, reducing miscarriage rates by 50% (29). Nevertheless, simpler techniques are available for selecting sperm with low DNA fragmentation such as PICSI and MACS, so IMSI-MSOME is not yet extendedly used (4). In fact, this technique did not have the expected impact due to its downsides: high cost and long time for selecting sperm for ICSI. Depending on semen quality, selection may take around 60-120 minutes, which could negatively affect cells given that sperm nuclei may vacuolize after 2-hour exposure to warm media (30). Polarized light microscope The different anisotropic properties of spermatozoa through its protoplasmic texture provoke polarized light to be refracted at different speeds. These differences between refractions are known as retardance or birefringence (31). Viable human spermatozoa are naturally birefringent, while this characteristic is absent in pathological, dead or necrotic ones due to changes in the molecular structures of the cell and/or organelles (nucleoprotein filaments, axoneme, mitochondria, etc.) (32, 33). Examined under polarized microscope (PM), a normal spermatozoon will exhibit a non-luminous acrosome and luminous and normal-sized compact nucleus and flagellum (Giulia Collodel 2010). Multiple studies support the correlation between sperm head birefringence and DNA fragmentation, fertilization rates and higher embryo quality (2). The increase of a sperm head retardance is positively correlated with DNA damage, the optimal value for sperm head retardance oscillates between 0.56 nm and 0.91 nm. (32). Thanks to improvements in polarized light microscopy it is now possible to evaluate viability, motility, morphology and concentration of a sperm sample, all in a single step and with no need of exposure to potentially harmful dyes or environmental conditions (33). MEMBRANE POTENTIAL BASED TECHNIQUES Zeta potential method Zeta potential refers to the negative electric potential observed between the outer medium and the sperm membrane surface. The sperm selection technique based on this potential was developed following the observation that sperm with damaged DNA exhibited a lower Zeta potential (34). This method consists in the use of positively charged test tubes to which morphologically normal spermatozoa containing intact DNA can bind (2). This promising, easy and time-efficient new method presents the major drawback of a low recovery rate (8.8%), which is especially troubling in cases of oligozoospermic patients, who represent a high percentage of ICSI candidates (34). Micro-electrophoresis During sperm maturation in the epididymis, capacitation and acrosome reaction, the sperm membrane undergoes specific modifications (35, 36), among which the addition of sialic acid residues is noteworthy. High concentrations of this residue reflect normal spermatogenesis and sperm maturation (37), as well as higher negative charge compared to immature or abnormal sperm (38). Taking advantage of such trait, negatively-charged spermatozoa are selected as they migrate towards the anode during electrophoretic sperm separation (39). Similarly to the Zeta-potential method, micro-electrophoresis enables the isolation of sperm with no DNA damage, since they present higher negative net charge (40). Although micro-electrophoresis remains an experimental method, the percentage of negatively charged sperm is directly associated with fertilization rate and blastocyst development and inversely associated with embryo arrest (41). ABSOLUTE IMMOTILE SPERM SELECTION TECHNIQUES: There are some cases of severe male factor infertility, such as total necrozoospermia, in which absolute immotile sperm are prevalent. In such cases, testicular sperm extraction (TESE) seems to be the only actual solution; however, obtaining motile and/or viable sperm for ICSI may result an impossible goal (42). Difficulties to select viable sperm can also occur in cases of severe cryptozoospermia and asthenozoospermia. Therefore, several techniques have been developed in order to differentiate immotile and non-viable sperm (2, 43, 44, 45). Hypo-osmotic swelling test (HOST) Studies have shown that in cases of severe male infertility normal sperm morphology will still likely have a high DNA fragmentation index. HOST is a method to estimate chromatin integrity (2), based on the degree of swelling of the cytoplasm and curling of the tail in live sperm when exposed to hypo-osmotic conditions. Different swelling patterns correspond to different degrees of chromatin integrity; by identifying these patterns, live spermatozoa with normal membrane function and low DNA fragmentation can be selected (2, 43). Laser assisted immotile sperm selection (LAISS) LAISS helps to differentiate between viable and dead spermatozoa by evaluating curling of the flagellum, as detected in live sperm when hit by the laser. The use of LAISS has reported results comparable to HOST; however, the former is much quicker than the latter, and its effect can be immediately observed without the need of continued evaluation, contrary to what is required for HOST (43). Mechanical touch technique or the sperm tail flexibility test (STFT) Although HOST is the most used test to differentiate live immotile spermatozoa from dead ones, evidence suggests that it may not be totally reliable (46, 47). Furthermore, solutions used for hypo-osmotic shock can be harmful for spermatozoa. In 2003, Soares and coauthors published their results using a simple and low-cost technique for selecting viable immotile sperm. They observed acceptable fertilization rates after injecting spermatozoa with flexible tail from total immotile sperm samples. Based on these results, the authors proposed STFT as a means to discriminate live from dead sperm; STFT consists in touching the flagellum with the ICSI pipette to check flexibility. Spermatozoa with flexible (non-stiff) flagellum may be considered alive and therefore eligible for ICSI (44). Methylxanthines: Pentoxifylline (PTX) Use of Pentoxifylline (PTX) enhances sperm motility by inhibiting the breakdown of cAMP which is essential for sperm motility (48). PTX is a 3’5’-nucleotide phosphodiesterase inhibitor that might result highly toxic for the oocyte or the embryo (2). Nevertheless, due to the reported increased fertilisation rates for immotile sperm after exposure to PTX (48), this method is still recommended for thawed testicular sperm samples, though only in cases of 100% immotile sperm, and always for short periods of time (2, 48). ATP/MgSO4 In 2014 Neri and colleagues proposed the use of ATP/MgSO4 solution for immotile spermatozoa that did not respond to motility enhancers (49). It is especially recommended for surgically retrieved or thawed sperm samples (49). The exposure to ATP/MgSO4 stimulate sperm kinetic machinery provoking viable, but immotile spermatozoa flagella to twitch (2). The same team reported that 64.6% of spermatozoa from an only immotile sperm sample exhibited flagellar movement after exposure to ATP/MgSO4 (49). CONCLUSIONS ICSI is a highly efficient technique, which reaches up to 85-90% fertilisation rates. Two main causes can explain fertilisation failure by this technique: the lack of appropriate sperm for injection or failed oocyte activation. Having covered the latter in a previous publication, the present post aims to review the current available techniques for the improved sperm selection for ICSI. Among these, IMSI, PICSI and MACS are widely studied, and their efficiency extensively accepted. Several novel approaches have been also developed. HOST, polarised microscopy and micro-electrophoresis are suggested to improve fertilisation rates, although multicenter randomised control trials will be needed before their general implementation to IVF routine. On the other hand, ZP binding assay and Zeta potential method are still experimental techniques that are yet to demonstrate their clinical advantages and feasibility. Finally, the best method for selecting live spermatozoa from totally immotile sperm samples remains a controversial issue. Whereas HOST seems to be the most extended one, simpler and easier techniques have appeared in the last decade that may also prove highly useful for an effective good-quality sperm selection. REFERENCES
Authors: Inés Abad, Roberto de la Fuente and Sara Sanz  Fig. 1. Close micrograph of a segment of the mid-piece of a mammalian spermatozoon, showing the helicoidally disposed mitochondria. (A) Cross section. (B) Surface of the plasma membrane. [modified from (1)]. INTRODUCTION Fertility treatments are more and more common in our days, reason why it is important to perform these procedures accurately resembling in vivo conditions. Additionally, male factor may oftentimes be underrated, and yet it is 50% of the treatment. The following text aims to establish an updated comparison between in vivo and in vitro semen preparation methods. In the first part a general description of the processes of maturation and capacitation of sperm are presented. MATURATION Where does sperm maturation take place? Once spermatogenesis is completed in the seminiferous epithelium, immature spermatozoa migrate towards the epididymis, the organ in which sperm maturation and storage take place. The epididymis is usually divided into three different parts: caput (head), corpus (body) and cauda (tail) (2). Typical changes in sperm during maturation 1. Acquisition of progressive motility. Even though immature sperm have functional movement machinery, motility of these cells begins in the caput segment. Whereas beating intensity is similar throughout the whole epididymis, flagellar amplitude is modified within this path. This is due to changes on the sperm surface, such as acquisition of new proteins and molecular changes involving inactivation of Ser/Thr phosphatases (3, 4). 2. Migration of the cytoplasmic droplet (CD). This droplet migrates from the neck towards the annulus of the mammalian spermatozoa (in humans, the CD is more proximal, located at the neck as opposed to the distal position of the annulus). The role of this droplet is to regulate ion homeostasis. It contains K+, Cl- and water channels, which have been suggested to work in regulation of sperm volume during the different regions of the epididymis. It also accumulates Ca2+, which has a biphasic role controlling phosphorylation pathways in sperm cells. In immature spermatozoa, it has been hypothesized that high Ca2+ levels found in the CD maintain low levels of tyrosine phosphorylation (5). 3. Changes in sperm protein and lipid profile. - Protein and lipid content Even though changes in these profiles are not well understood, there are three complementary mechanisms that participate in completing maturation:
Perhaps the most important among these changes is the significant reduction in cholesterol content of the sperm membrane. This reduction involves a decrease in the cholesterol/phospholipid ratio that facilitates protein trafficking from and onto the membrane and enhances its fluidity, which will eventually play a role in triggering capacitation and fertilisation (8). - Post-translational protein changes Additionally, certain post-translational modifications of proteins have also been hypothesised to occur during sperm maturation. This is the case for oxidation of thiol groups, which promotes the formation of disulphide bonds (S-S) and stabilises components of both the head and flagellum (9). 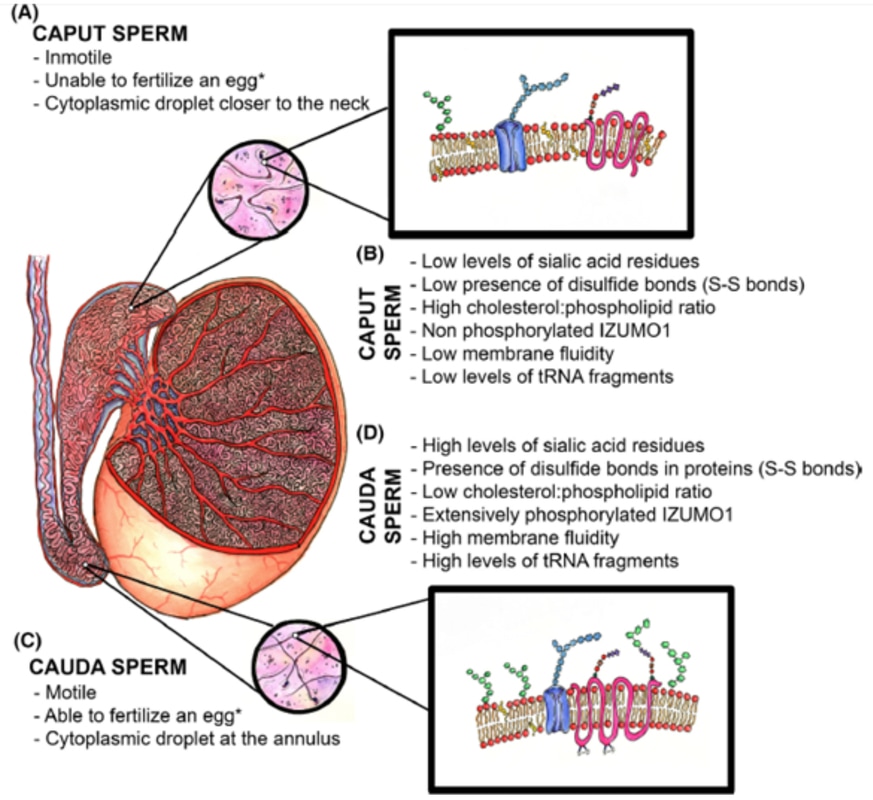 Fig.2. Schematic representation of the main items during sperm maturation. (A) Principal functional and morphological aspects in immature caput spermatozoa. (B) Molecular characteristics of immature spermatozoa. (C) Main morphofunctional traits of cauda mature spermatozoa and (D) their molecular features (4) (*) Ability to fertilize the egg will ultimately depend on completing capacitation. CAPACITATION What is the composition of the ejaculate? In mammals, semen is composed of two different phases:
Following ejaculation, semen is deposited to the anterior wall of the vagina, adjacent to the ectocervical tissues. From here on, for sperm to progress towards the egg through the oviduct or Fallopian tubes, semen must undergo liquefaction. This process usually takes about 20-30 minutes (11). Semen goes through the cervix and reaches the distal portion of the Fallopian tube, where sperm is stored and maintained by interacting with the endosalpingeal epithelium (12, 13). In 1951, Austin and Chang individually observed that a certain period of time in the female tract was required for sperm before fertilization could take place (14, 15). Later, observations in multiple mammalian species confirmed these first notions, and certain studies showed a delay of at least 2 hours before sperm entry into the egg. This supports the hypothesis of sperm maturation before becoming fertile after ejaculation (16). Nowadays, such process is known as capacitation, as opposed to maturation in the male tract explained above. Capacitation and fertility Some of the factors involved in sperm capacitation are steroid hormones such as oestrogens and progesterone, both produced by the follicle. These steroids play different roles: they act as chemoattractants, facilitate triggering of hyperactivation, regulate trafficking of cGMP or modulate the potential for completing acrosome reaction (17-19). Semen liquefaction following ejaculation is mainly modulated by prostate derived peptidase KLK3. In females, KLKs 5–8, 10–11, and 13–15 are expressed at very high levels in the cervix and vagina compared to other adult tissues (20, 21). Moreover, KLK1 and KLK3 transcripts are expressed at the highest level in human endometrium when circulating estradiol (E2) is elevated. These findings suggest that KLKs are expressed in the human reproductive tracts and that some of the KLKs in the uteri are regulated by E2. Abnormal E2 signalling in the female reproductive tract leads to semen liquefaction defects, associated with defective SEMG cleavage and sperm transport, which may result in some cases of infertility. It is known that mice lacking ESR1 (one of the oestrogen receptors) in the epithelial cells are infertile (22), partly due to a reduction in the number of sperm able to reach the oviduct (23). However, the effect of ESR1 loss in the epithelial cells on sperm transport in the uterus has not yet been investigated. Similarly, other potential research lines could investigate liquefaction defects caused by diminished KLK activity in females or regulation of KLKs by molecular signalling in the female tract. Once semen trespasses the cervix, sperm are known to achieve capacitation in an asynchronous fashion during the interaction with the epithelium, which results in a continuous flow of fertile spermatozoa towards the Graafian follicle (24). The ability to bind to the epithelium, in turn, may be indirectly related to the sperm DNA integrity, and so DNA fragmentation levels would be indicative of the fertility potential of the sperm (25). Elements involved in sperm capacitation Even though capacitation had traditionally been regarded as a two-step process, through which changes in the cell membrane would lead to the acrosome reaction (AR) (26), capacitation is currently considered as a continuous process that culminates in the AR. It would be difficult to describe all capacitation-related events separately because all of them are connected to each other in time. However, the most important changes in the sperm during the process are (27-37):
Sperm capacitation is a complex process with multitude of interconnected and highly regulated molecular pathways. One of the first events is the alteration of the permeabilization of the sperm plasma, so that the influx and intracellular concentration of certain ions are increased. The main molecules involved are probably Ca2+ and HCO3-; the net intake by the sperm cell triggers alkalinisation of the pH and the concomitant activation of the soluble adenylyl cyclase (sAC) (38, 39). As an immediate consequence, cAMP levels increase followed by activation of the protein kinase A (PKA) (40, 41). The rise of cAMP causes redistribution of certain phospholipids and proteins of the membrane, and so exposing cholesterol, which accumulates in lipid rafts (42). The organization of these rafts promotes the removal of cholesterol and its translocation to extracellular acceptors like albumin (43). Also, increased cAMP activates PKA, which in turn activates SRC kinase (44). Eventually, SRC kinase activity triggers tyrosine phosphorylation, which results and a wide range of proteins been modified and relocated in capacitating sperm. This has been described in several species, including humans (45). The end result of capacitation is the acrosome reaction (AR), the process by which the content of the acrosome is released to the extracellular environment. In natural conditions, this environment is actually the cummulus cells, whose connections will be broken by the chemical reactions of the acrosomal content, mainly proteases like acrosin and hyaluronidase, also exposed to the right membrane domains during lipid redistribution (46). It is not surprising that mutations affecting any of these processes will result in multiple causes for infertility (47). FUTURE PERSPECTIVES Findings like the one regarding post-ejaculated liquefaction, mutations on acrosome protease-encoding genes or other molecular mechanisms of sperm capacitation are crucial to progress in the field of reproductive medicine, and can lead to: (i) potential diagnostic tools for unexplained infertility cases, (ii) the development of a novel contraception technology to entrap sperm (48), (iii) or even revolutionary new methods for human sperm capacitation in the laboratory (49), which could significantly improve live birth rates for fertility treatments. In the following post... different methods for sperm selection in the laboratory will be explained, paying attention to advantages and disadvantages under different circumstances. The importance of different sperm features like DNA fragmentation or morphology will be discussed in relation to the best sperm selection method to achieve optimal clinical outcomes. REFERENCES:
|
Entries
March 2019
Categories
All
 2016-2019. All Rights Reserved by Embryologist Media. This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License . |
Embryologist Media
